Laste ned presentasjonen
Presentasjon lastes. Vennligst vent

1
Et trygt og robust samfunn – der alle tar ansvar MTF – LANDSMØTE TROMSØ 28. MAI 2009 DE NYE FORSKRIFTENE ???

4
Gebyrforskriften FEF FEL/NEK Forskrift om elektrovirksomhet (FKE) Forskrift om elektrisk utstyr (FEU) Forskrift om medisinsk utstyr
 Forskrift om elektrisk utstyr (FEU) Forskrift om medisinsk utstyr")
5
Gebyrforskriften Er tilbehandling i departementet

6
FEF Tilbakevirkende kraft bl.a.: Høyspenning kutter ved første jordfeil Konsekvens: Helseforetakene må større grad stole på egen strømforsyning.

7
FEL/NEK (ny i 2010) Sannsynlige endringer bl.a.: Krav til ledningsresistanse i gruppe 1 rom – 0,7 Plassering av uttak medisinske gasser Krav til brannceller(UPS-generatorer- hovedtavler)
 Sannsynlige endringer bl.a.: Krav til ledningsresistanse i gruppe 1 rom – 0,7 Plassering av uttak medisinske gasser Krav til brannceller(UPS-generatorer- hovedtavler)")
8
Forskrift om elektrovirksomhet (nå FKE) Til behandling i departementet pga Tjenestedirektivet
 Til behandling i departementet pga Tjenestedirektivet")
9
Direktivene del av ”New Approach” Ny Metode: Ny Metode: fri flyt av varer og tjenester, ikke handelshinder fri flyt av varer og tjenester, ikke handelshinder konsekvens av EØS-avtalen konsekvens av EØS-avtalen ”Gammel Metode” ”Gammel Metode” nasjonale godkjenningsordninger nasjonale godkjenningsordninger

10
Forskrift om elektrisk utstyr (FEU) Virkning fra 31. oktober 2008 Lavspenningsdirektivet EMC direktivet (veterinærdirektivet)
.")
13
Forskrift om medisinsk utstyr

15
Grunnleggende krav (bl.a.) Sikkerhet Risiko unngått eller begrenset Ytelser og egenskaper som angitt Konstruksjon og produksjon Kan antas oppfylt om produsenten har fulgt bestemte standarder (= harmoniserte standarder)
 Sikkerhet Risiko unngått eller begrenset Ytelser og egenskaper som angitt Konstruksjon og produksjon Kan antas oppfylt om produsenten har fulgt bestemte standarder (= harmoniserte standarder)")
16
Teknisk dokumentasjon sikkerhet, ytelse, bruksområde konstruksjon, virkemåte, fremstiling, testing, risikoanalyse, kliniske evalueringer,... Dokumentasjonen skal gjøre det mulig å vurdere om produktet er i samsvar med de grunnleggende krav i direktivene.

17
Samsvarsvurdering Produsenten skal utarbeide den tekniske dokumentasjon, …..skal gjøre det mulig å vurdere om produktet er i samsvar med kravene i dette direktiv. Produsent vurderer opp mot direktivets krav,.... garanterer og erklærer at de aktuelle produktene oppfyller de bestemmelser i dette direktiv som gjelder for dem.....,...skal påføre CE-merking i samsvar med artikkel... Er basert på ulike prosedyrer avhengig av den risiko som er forbundet med utstyret dersom det svikter

18
Risikoklassifisering Avgjør hvilken vurderingsprosedyre som skal benyttes Klassifiseringen avspeiler: risikoen som er forbundet med bruk, sårbarheten av de legemsdelene utstyret skal brukes på, og varigheten av bruken Høyeste klasse (III) omfatter bl.a. produkter som kommer i kontakt med sentralnervesystemet, hjertet og det sentrale kretsløp, samt medisinsk utstyr som inneholder legemidler

19
Teknisk kontrollorgan Uavhengige, faglig kompetente foretak Utpekt av myndighetene til å vurdere produkter og/eller kvalitetssystemer Involvert ved samsvarsvurdering for alle produkter unntatt individuelt tilpasset og klasse I; klasse I (når steril eller målefunksjon) klasse IIa klasse IIb klasse III
 klasse IIa klasse IIb klasse III")
20
EFs samsvarserklæring Produsenten skal alltid utstede en erklæring om at hans produkter oppfyller direktivets krav før CE-merking kan påføres. Erklæringen samt teknisk dokumentasjon skal oppbevares av produsenten og på anmodning gjøres tilgjengelig for fagmyndighet og/ eller teknisk kontrollorgan

21
CE-merking Synlig tegn på at utstyret er i tråd med regelverket Grunnleggende krav er oppfylt Samsvarsvurdering er foretatt

22
§ 1-3 (unntak fra forskriftens virkeområde) – e) Utstyr som produseres og brukes i)Av helseinstitusjonen som har produsert det, eller i lokaler i umiddelbar nærhet av produksjonsstedet uten å utgjøre en annen juridisk enhet enn dette, og ii) til helseinstitusjonens ordinære arbeidsoppgaver, og iii) uten at utstyret utnyttes kommersielt For elektromedisinsk utstyr er ”in-house” produksjon regulert i forskrift om bruk og vedlikehold av elektromedisinsk utstyr Innledende bestemmelser NB !! (In-house)
.")
23
Forskriften retter seg i hovedsak mot produsenter og leverandører. Det er spesielt to forhold som vil berøre sykehuset: §2-7 at utstyret oppbevares og lagres i samsvar med produsentens spesifiseringer. § 2-12 Plikt til å melde hendelser der elektromedisinsk utstyr har vært eller kan ha vært innvolvert. Bruk av elektromedisinsk utstyr dekkes av forskrift om bruk og vedlikehold av elektromedisinsk utstyr. Dagens forskrift og ny forskrift retter seg mot å sikre at utstyr på markedet er i samsvar med de harmoniserte regler Tilsynsmyndigheten kan fastsette retningslinjer for meldeplikten

24
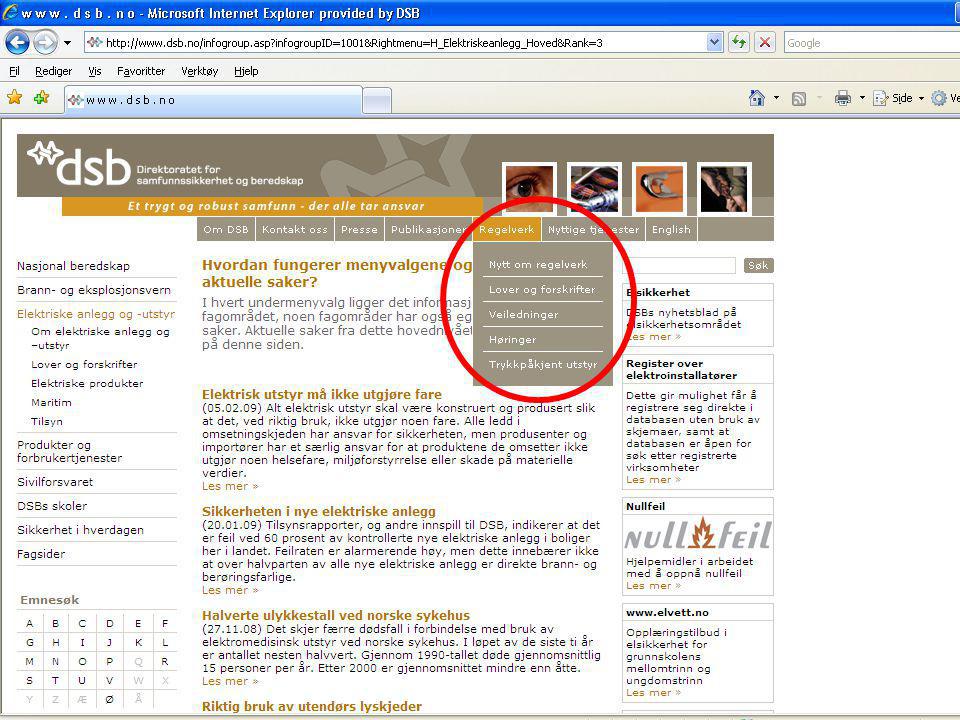
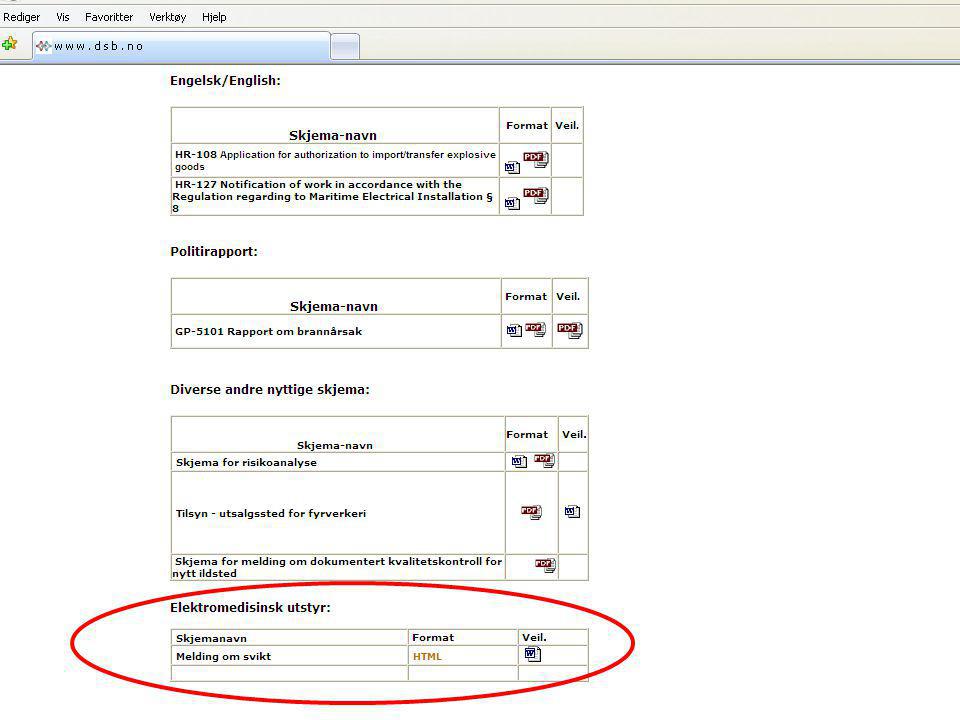
Den som i sin virksomhet eier eller bruker utstyr, plikter uten unødig opphold å melde om hendelser som har eller kunne ha ført til død eller betydelig skade på pasient, bruker eller annen person og som har eller kan ha sammenheng med bruk av utstyret. Vakttelefon 48 21 20 00 Mindre alvorlige ”hendelser” meldes telefonisk første virkedag etter ”hendelsen”. Telefon 33 41 25 00 Forskrift om medisinsk utstyr. Eller bruk fellesskjema til H-dir./DSB VI. Meldeplikt; § 2-12. (brukers og eiers plikt til å melde)
.")
30
Meldeplikt for produsenter og omsettere § 2-11. (plikt til å melde uhell mv.) Den som i sin virksomhet produserer eller omsetter utstyr, skal uten unødig opphold melde om: enhver feilfunksjon eller enhver forringelse av et utstyrs egenskaper og/eller ytelser samt enhver mangel på merkingen eller bruksanvisningen som kan føre til eller kunne ha ført til pasientens, brukerens eller annen persons død eller alvorlig forverring av pasientens, brukerens eller annen persons helsetilstand, a)
 Den som i sin virksomhet produserer eller omsetter utstyr, skal uten unødig opphold melde om: enhver feilfunksjon eller enhver forringelse av et utstyrs egenskaper og/eller ytelser samt enhver mangel på merkingen eller bruksanvisningen som kan føre til eller kunne ha ført til pasientens, brukerens eller annen persons død eller alvorlig forverring av pasientens, brukerens eller annen persons helsetilstand, a).")
31
ethvert teknisk eller medisinsk hensyn i forbindelse med et utstyrs egenskaper eller ytelser som, av de årsaker som er omhandlet under bokstav a), har ført til at produsenten systematisk har trukket utstyr av samme type tilbake fra markedet eller gjort endringer som følge av dette. b) Meldingen sendes tilsynsmyndigheten.
 Meldingen sendes tilsynsmyndigheten..")
32
Et system for landene i EØS-området + Sveits. Ikraftreden 01.01.08 (har vært en frivillig ordning frem til da) Gjelder utstyr som dekkes av direktivene i forskrift om medisinsk utstyr. Brukerne skal melde hendelser til myndighetene DSB ev. Hdir. Myndighetene melder til fabrikant. Myndighetene overvåker fabrikant og ev. iverksetter ytterligere tiltak for å supplementere fabrikantens tiltak. Myndighetene skal gi nødvendig informasjon for å hindre ytterligere hendelser. Fabrikant har ansvaret for å ”etterforske” hendelsen og iverksette tiltak. Overvåkningssystem for medisinsk utstyr, Viglance
 Gjelder utstyr som dekkes av direktivene i forskrift om medisinsk utstyr. Brukerne skal melde hendelser til myndighetene DSB ev. Hdir. Myndighetene melder til fabrikant. Myndighetene overvåker fabrikant og ev. iverksetter ytterligere tiltak for å supplementere fabrikantens tiltak. Myndighetene skal gi nødvendig informasjon for å hindre ytterligere hendelser. Fabrikant har ansvaret for å etterforske hendelsen og iverksette tiltak. Overvåkningssystem for medisinsk utstyr, Viglance.")
33
Takk for meg!

Liknende presentasjoner