Laste ned presentasjonen
Presentasjon lastes. Vennligst vent

1
MBV3070 Bioinformatikk; proteiner
Lærer: Vincent Eijsink, Institutt for Kjemi, Bioteknologi og Matvitenskap, Norges landbrukshøgskole; Telefon: ; E-post: Web: kitin.nlh.no/enzymgruppa Pensum: Tatt fra Lesk, side 31-53, , og , med fokus På kapittel 5 (s ) og utvidet med tilleggsinformasjon fra andre kilder. Pensumet består av: Det som behandles under forelesningene (Lysbildene) Øvelsene Guex et al. Protein modelling for all TIBS 24: (1999) (om SwissModel) Schonbrun et al., Protein structure prediction in 2002, Current Opinion in Structural Biology, 12: (2002) Les boka! Det er bra skrevet.
 og utvidet med tilleggsinformasjon fra. andre kilder. Pensumet består av: Det som behandles under forelesningene (Lysbildene) Øvelsene. Guex et al. Protein modelling for all TIBS 24: (1999) (om SwissModel) Schonbrun et al., Protein structure prediction in 2002, Current Opinion in Structural Biology, 12: (2002) Les boka! Det er bra skrevet.")
2
MBV3070 Bioinformatikk; proteiner
Program: Ti 20.04: forelesninger Fre 30.04: forelesninger Ti : forelesninger; øvelsene deles ut Fre 07.05: forelesninger; diskusjon av de artikler som er pensum Ti 18.05: gjennomgang av øvelsene; spørsmål, oppsummering

3
Tema Protein struktur og folding, oversikt (repetisjon)
Strukturbestemmelse av proteiner Protein struktur databaser (PDB og andre) Visualisering av strukturer Sammenligning og kategorisering av av strukturer Multiple sequence alignments (MSAs) Databaser av MSAs Sekvens-basert prediksjon av funksjon, evolusjonær opphav, struktur, fysiske egenskaper Fold recognition (”threading”) Prediksjon av sekundær og tertiær struktur; bygging av tre-dimensjonale protein modeller Proteomics, structural genomics Bruk av proteinkunnskap i annotering av genomer Avansert protein bioinformatikk (f eks molekylær dynamika, elektrostatika, protein design – stort sett bare eksempler)
 Visualisering av strukturer. Sammenligning og kategorisering av av strukturer. Multiple sequence alignments (MSAs) Databaser av MSAs. Sekvens-basert prediksjon av funksjon, evolusjonær opphav, struktur, fysiske egenskaper. Fold recognition ( threading ) Prediksjon av sekundær og tertiær struktur; bygging av tre-dimensjonale protein modeller. Proteomics, structural genomics. Bruk av proteinkunnskap i annotering av genomer. Avansert protein bioinformatikk (f eks molekylær dynamika, elektrostatika, protein design – stort sett bare eksempler)")
4
Generelt om proteiner Matthews, Van Holde & Ahern (KJB201) eller andre generelle biokjemi tekstbøker C. Branden & J. Tooze, Introduction to Protein Structure (ISBN ) A. Lesk, Introduction to Protein Science (ISBN ) G.A. Petsko, D. Ringe, Protein Structure and Function (ISBN: )
 A. Lesk, Introduction to Protein Science (ISBN ) G.A. Petsko, D. Ringe, Protein Structure and Function (ISBN: )")
5
Andre nyttige bioinformatikk bøker
Mount, D., Bioinformatics – Sequence and genome analysis ( Baxevanis, A.D. & Ouellette, B.F., Bioinformatics. A practical guide to the analysis of genes and proteins De som vil vite mer om model-bygging kan lese: Rodriguez & Vriend, ”Professional gambling”, (en detaljert og relativ enkel forklaring av prinsippene av ”model-building- by- homology” som programmet WHAT IF gjør det)
")
6
modellering av protein struktur?
Bioinformatikk = modellering av protein struktur? Se C. Levinthal, Scientific American, juni 1966: ”Molecular Model-building by computer” (ikke pensum)
")
7
INTRODUKSJON

8
Protein struktur; terminologi
Primærstruktur: Aminosyrerekkefølge Sekundærstruktur: Faste strukturelementer, karakterisert av standard type hydrogen bindinger mellom hovedkjede atomer: a-helix, b-sheet, turn, coil mfl. (ofte deler man i bare tre varianter: a-helix, b-sheet, rest) Tertiær struktur: Hele strukturen, inklusive alle detaljer (= ”tertiære interaksjoner”) Kvarternær struktur: multimere proteiner; hver subunit har sin tertiær struktur; hele komplekset har en kvarternær struktur
 Tertiær struktur: Hele strukturen, inklusive alle detaljer (= tertiære interaksjoner ) Kvarternær struktur: multimere proteiner; hver subunit har sin tertiær struktur; hele komplekset har en kvarternær struktur.")
9
Amino acids OBS! Bilder av proteinstrukturer viser som
oftest ikke hydrogen atomer

10
Amino acids

11
Amino acids Det finnes mange måter for å gruppere aminosyrer på, og dette er ikke triviell (jfr f eks sekvenssammenligning)
")
12
Alanine Oksygen Nitrogen Karbon Hydrogen Glycine (R = H) og proline
(R kovalent bundet til N) er spesial
 er. spesial.")
13
Stereokjemi: CORN-regelen
All amino acids found in proteins encoded by the genome have the L-configuration at this chiral centre. This configuration can be remembered as the CORN law. When read clockwise, the groups attached to the Calpha spell the word CORN.

14
Peptidbindingen

15
Peptidbindingen er plan
O O- - N – C N = C - H H H C-a C-a N As a consequence of this resonance all peptide bonds in protein structures are found to be almost planar, ie atoms Calpha(i), C(i), O(i), N(i+1) H(i+1) and Calpha(i+1) are approximately co-planar. This rigidity of the peptide bond reduces the degrees of freedom of the polypeptide during folding. H R H
, C(i), O(i), N(i+1) H(i+1) and Calpha(i+1) are approximately co-planar. This rigidity of the peptide bond reduces the degrees of freedom of the polypeptide during folding. H. R. H.")
16
Rotasjonsakser i peptider
H H H y f N C-a C-a C-a N H R H Phi og Psi begrenses av generell kjemiske prinsipper (”staggered” er bedre enn ”eclipsed”); variasjon i R påvirker mulighetene.
; variasjon i R påvirker mulighetene.")
17
Torsion Angles Ingen vesentlig rotasjon om omega.
Bindingsvinkler og lengder viser ingen vesentlig variasjon Phi & Psi avgjør strukturen

18
Ramachadran plot Ramachadranplott viser kombinasjoner av f og y som fore-kommer i protein strukturer Dette er dermed ener-getisk gunstige kombi-nasjoner av f og y I slike plots skiller glysin og proline seg ut In the diagram above the white areas correspond to conformations where atoms in the polypeptide come closer than the sum of their van der Waals radi. These regions are sterically disallowed for all amino acids except glycine which is unique in that it lacks a side chain. The red regions correspond to conformations where there are no steric clashes, ie these are the allowed regions namely the alpha-helical and beta-sheet conformations. The yellow areas show the allowed regions if slightly shorter van der Waals radi are used in the calculation, ie the atoms are allowed to come a little closer together. This brings out an additional region which corresponds to the left-handed alpha-helix.

19
Side Chain Conformation

20
Sekundær struktur (side 40, Fig. 1.7)
Strukturelementer med karakteristiske mønstre med hydrogenbruer: alpha-helix 310 helix b-flak (sheet, strand) Diverse typer ”turns” Ofte snakker man bare om 3 ”states”: helix, strand, coil
 Diverse typer turns Ofte snakker man bare om 3 states : helix, strand, coil.")
21
Properties of the alpha-helix
Regelmessig hydrogenbru mønster ”Overskudd” av N-H på N-terminus og av C=O på C-terminus -> Dipol

22
a-helix

23
En a-helix er ofte amfipatisk:
Helical wheel

24
b-strand Sidegruppene peker vekselvis ut på hver side av beta-flaket; en enkel beta-strand kan også være amfipatisk En beta-”strand” har karakteristiske phi og psi vinkler. Flere beta-strands kan gå sammen og danne en beta-”sheet”

25
b-sheet types

26
Beta sheets are usually twisted

27
b-sheet

28
Vesentlig forskjell mellom a og b:
Helikser stabiliseres av interaksjoner som er ”lokal” i sekvensen. b-sheets stabiliseres av interaksjoner mellom hovedkjedeatomer som kan ligge langt fra hverandre i sekvensen (Hvilke av disse to typer struktur vil være letter å predikere på basis av kun aminosyresekvensen?)
")
29
“Supersekundærstruktur” (“motifs”)
Kombinasjoner av påfølgende sekundær-strukturelementer (SSEer), f eks bab, b-hårnål, aa, b-tønne
, f eks bab, b-hårnål, aa, b-tønne.")
30
Helix-turn-helix

31
b-tønne Cellulært retinolbindende protein

32
(ba)8 barrel Cellulært retinolbindende protein
8 barrel Cellulært retinolbindende protein")
33
Protein struktur og folding
For å bli til et funksjonelt protein må den nysyntetiserte aminosyrekjeden få en tertiær struktur. Denne strukturen må være: Tilstrekkelig stabil Oppnåelig Oppnåelig vil si: Det må finnes en folding ”pathway” som fører til at strukturen blir dannet ”Off-pathway” prosesser (f eks utfelling / aggregering) må motarbeides
 må motarbeides.")
34
Chaperones (ikke pensum)
Chaperoner og chaperoniner beskytter mot ”off-pathway” prosesser, f eks GroEL/ES systemet [bildet er fra Science 284: (1999)]
]")
35
Protein folding (side 224-225)
The Levinthal paradox is more or less solved: Local (secondary) interactions and an intrinsic tendency of unfolded proteins to form (transient) elements of secondary structure steer the protein into a ”folding funnel”. D. Baker, Nature 405:39-42 (2000)
 interactions and an intrinsic tendency of unfolded proteins to form (transient) elements of secondary structure steer the protein into a folding funnel . D. Baker, Nature 405:39-42 (2000)")
36
Protein folding (side 224-225)
Three steps: Transient elements of secondary structure Hydrophobic collapse, sekundær struktur dannelse Completion of folding (tertiary interactions) The rate-limiting step is between phase 2 and 3 and is the same for all molecules Names for this mechanism: ”nucleation condensation” (Fersht), ”extended nucleus”, framework model (Kim & Baldwin)
 The rate-limiting step is between phase 2 and 3 and is the same for all molecules. Names for this mechanism: nucleation condensation (Fersht), extended nucleus , framework model (Kim & Baldwin)")
37
Protein struktur og stabilitet (side 221- 224)
For (nesten) hvert protein finnes det en unik struktur som er den energetisk mest gunstige. Stabilitet er marginal og er summen av store negative effekter og store positive effekter av folding Negativ effekt av folding: tap av entropi Positiv effekt av folding: hydrofob effekt Det finnes mange ”restraints” som begrenser hva som er mulig i naturen (se side ; eksempel: Ramachandran plott)
 hvert protein finnes det en unik struktur som er den energetisk mest gunstige. Stabilitet er marginal og er summen av store negative effekter og store positive effekter av folding. Negativ effekt av folding: tap av entropi. Positiv effekt av folding: hydrofob effekt. Det finnes mange restraints som begrenser hva som er mulig i naturen (se side ; eksempel: Ramachandran plott)")
38
PROTEIN STRUKTUR: Eksperimentelle metoder Databaser Visualisering

39
Eksperimentell bestemmelse av proteinstruktur
Lav-resolusjons teknikker: Circulær dikroisme sepktroskopi (sekundær struktur, stort sett ”all-or-nothing”) Fluorescence (tertiær struktur – ”all-or-nothing”) Elektronmikroskopi (brukes av og til for å få et lav-resolusjonsbilde av store proteinkomplekser) Röntgenkrystallografi Proteinet må krystalliseres Elektrontettheten bestemmes Atomkoordinatene utledes Gir statiske modeller Bottle neck: krystallisering NMR Analyserer kjernespinnsresonnans Utleder (flere) modeller som er konsistente med resonnansmønsteret Viser reell variasjon for peptider i løsning, men også alternative modeller man ikke kan skille mellom Bottle neck: complexity, solubility, labour power (but things are improving!)
 Fluorescence (tertiær struktur – all-or-nothing ) Elektronmikroskopi (brukes av og til for å få et lav-resolusjonsbilde av store proteinkomplekser) Röntgenkrystallografi. Proteinet må krystalliseres. Elektrontettheten bestemmes. Atomkoordinatene utledes. Gir statiske modeller. Bottle neck: krystallisering. NMR. Analyserer kjernespinnsresonnans. Utleder (flere) modeller som er konsistente med resonnansmønsteret. Viser reell variasjon for peptider i løsning, men også alternative modeller man ikke kan skille mellom. Bottle neck: complexity, solubility, labour power (but things are improving!)")
40
Experimentell struktur bestemmelse
Gir oss strukturdatabasen som er grunnlaget for mange prediksjonsmetoder Tar mye mer tid enn ”bioinformatisk struktur bestemmelse” (f. eks. modell bygging) Men ”bioinformatisk struktur bestemmelse” er ikke alltid mulig og gir dessuten mindre nøyaktige resultater
 Men bioinformatisk struktur bestemmelse er ikke alltid mulig og gir dessuten mindre nøyaktige resultater.")
41
Hvordan beskriver man en struktur?
Kjemisk sammensetning Forbindelser mellom atomene (”chemical connectivity”) Atom koordinater, x, y og z Vann molekyler og ligander Chemical bonds in structures: Chemistry rules approach: man bruker kjemiske regler for å rekonstruere bindinger i et bilde Explicit bonding approach: all informasjon om ”bonds” ligger i koordinatfilen Nb. ”Completeness”: strukturfiler er som oftest ikke komplett: Små deler av proteinet mangler; i nesten alle krystalstrukturer har man ikke protoner med
 Atom koordinater, x, y og z. Vann molekyler og ligander. Chemical bonds in structures: Chemistry rules approach: man bruker kjemiske regler for. å rekonstruere bindinger i et bilde. Explicit bonding approach: all informasjon om bonds ligger i koordinatfilen. Nb. Completeness : strukturfiler er som oftest ikke komplett: Små deler av proteinet mangler; i nesten alle krystalstrukturer. har man ikke protoner med.")
42
Mer avansert søk

43
Stadig flere strukturer -> en stadig bedre database for å
oppnå forståelse og for å utvikle prediksjonsmetoder OBS! Mange ”redundant” structures……………

44
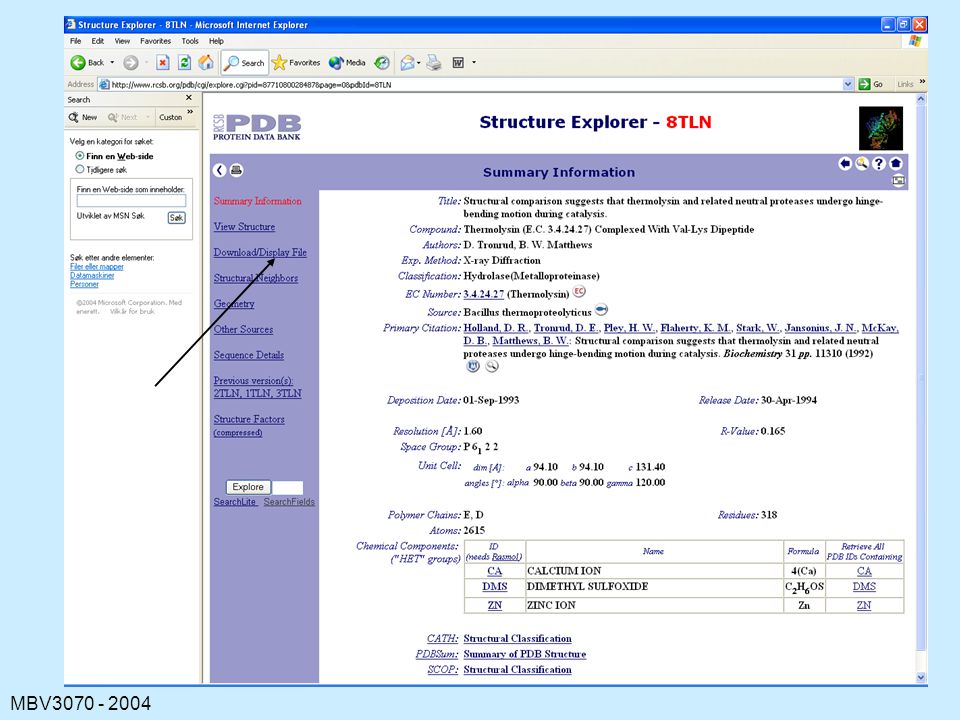
Søk på 8tln

47
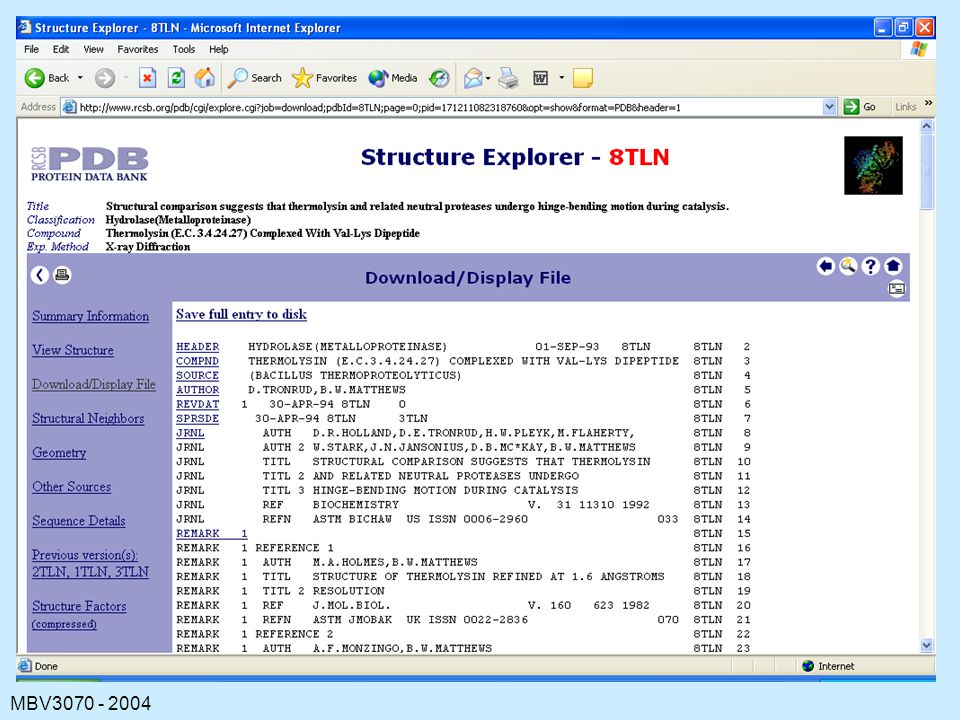
The Contents of a PDB File
HEADER: containing the file name and date. TITLE: usually of a publication COMPND: containing the name of the protein. SOURCE: the organism from which the protein was obtained. KEYWORDS EXPDTA: method used AUTHORS: persons who placed this data in the PDB REVDAT: revision dates for data on this protein. JRNL: relevant publications REMARK: various types of information about the experiments, the file, symmetry, missing residues, quality checks (REMARK is usually many lines)
")
48
The Contents of a PDB File
DBREF: accession codes for this protein in other databases SEQRES: explicit amino-acid sequence of the protein. HET, HETNAM, FORMUL; information on cofactors, prosthetic groups, inhibitors or other nonprotein substances present in the structure. HELIX, SHEET: elements of secondary structure in the protein. LINK: contacts between heteroatoms and amino acids CISPEP: peptide bonds in cis SITE: information of binding sites and active sites CRYST, ORIGX, SCALE: technical information on the coordinates, symmetry operations ATOM and HETATM: atomic coordinate data

49
The Contents of a PDB File
SEQRES VAL ASN CYS ALA LYS LYS ILE VAL SER ASP GLY ASN GLY 1HEW 67 SEQRES MET ASN ALA TRP VAL ALA TRP ARG ASN ARG CYS LYS GLY 1HEW 68 SEQRES THR ASP VAL GLN ALA TRP ILE ARG GLY CYS ARG LEU HEW 69 HET NAG N-ACETYL-D-GLUCOSAMINE HEW 70 HET NAG N-ACETYL-D-GLUCOSAMINE HEW 71 HET NAG N-ACETYL-D-GLUCOSAMINE HEW 72 FORMUL 2 NAG 3(C8 H15 N1 O6) HEW 73 FORMUL 3 HOH *103(H2 O1) HEW 74 HELIX A ARG HIS HEW 75 HELIX B LEU GLU HEW 76 HELIX C CYS LEU HEW 77 HELIX D THR ILE HEW 78 HELIX E VAL ASN HEW 79 SHEET S1 2 LYS PHE HEW 80 SHEET S1 2 PHE THR N THR O LYS HEW 81 OBS! Assignment av sekundær struktur er ikke triviell.
 1HEW 73. FORMUL 3 HOH *103(H2 O1) 1HEW 74. HELIX 1 A ARG 5 HIS HEW 75. HELIX 2 B LEU 25 GLU HEW 76. HELIX 3 C CYS 80 LEU HEW 77. HELIX 4 D THR 89 ILE HEW 78. HELIX 5 E VAL 109 ASN HEW 79. SHEET 1 S1 2 LYS 1 PHE 3 0 1HEW 80. SHEET 2 S1 2 PHE 38 THR N THR 40 O LYS 1 1HEW 81. OBS! Assignment av sekundær struktur er ikke triviell.")
50
The Contents of a PDB File
The number and type of items/subjects in the header may vary. The PBD keeps the actual X-ray data, the BioMagRes bank (Wisconsin) keeps the data for NMR structures Keep in mind that the entry names (4 characters) are not ”logical” (that is usually not like e.g. ”8tln” for thermolysin) See also in the book, page 125 – 131.
 keeps the data for NMR structures. Keep in mind that the entry names (4 characters) are not logical (that is usually not like e.g. 8tln for thermolysin) See also in the book, page 125 – 131.")
51
Atomic coordinates in PDB files
X Y Z B ATOM HA CYS BBN 897 ATOM HB CYS BBN 898 ATOM HB CYS BBN 899 ATOM N ARG BBN 900 ATOM CA ARG BBN 901 ATOM C ARG BBN 902 ATOM O ARG BBN 903 ATOM CB ARG BBN 904 ATOM CG ARG BBN 905 ATOM CD ARG BBN 906 ATOM NE ARG BBN 907 ATOM CZ ARG BBN 908 ATOM NH1 ARG BBN 909 ATOM NH2 ARG BBN 910 Note that hydrogen atoms are not listed. Almost all protein crystals do not diffract well enough to allow hydrogen atoms to be resolved. Positions of hydrogen atoms must be inferred from the positions of other atoms. B-factor: says something about how well the position was defined by the electron density (-> an indicator of mobility and accuracy) a) atom number: atoms are numbered in sequence through the file; b) atom type: n = amide N, ca = alpha C, c = carbonyl C, o = carbonyl O, cb = beta carbon, and so forth; c) residue name: three-letter amino acid abbreviation; d) residue number; e) x-coordinate of the atom, in angstroms from the unit-cell origin; f) y-coordinate of the atom; g) z-coordinate of the atom; h) occupancy: the fraction of unit cells that contain the atom in this particular location, usually 1.00, or all of them (can be used to represent alternative conformations of side chains); i) temperature factor: an indication of uncertainty in this atom's position due to disorder or thermal vibrations (can be used by graphics programs to represent the relative mobility of different parts of a protein) j) every line ends with the PDB file identification code.
 a) atom number: atoms are numbered in sequence through the file; b) atom type: n = amide N, ca = alpha C, c = carbonyl C, o = carbonyl O, cb = beta carbon, and so forth; c) residue name: three-letter amino acid abbreviation; d) residue number; e) x-coordinate of the atom, in angstroms from the unit-cell origin; f) y-coordinate of the atom; g) z-coordinate of the atom; h) occupancy: the fraction of unit cells that contain the atom in this particular location, usually 1.00, or all of them (can be used to represent alternative conformations of side chains); i) temperature factor: an indication of uncertainty in this atom s position due to disorder or thermal vibrations (can be used by graphics programs to represent the relative mobility of different parts of a protein) j) every line ends with the PDB file identification code.")
52
Eksplisite og implisite sekvenser
Eksplisitt sekvens (SEQRES): sekvensen av hele proteinet Implisitt sekvens: sekvensen som ligger i strukturfilen Siden strukturfiler ofte mangler noen residuer (f eks på N-terminus) er implisitt og eksplisitt sekvens ofte ikke like. Dette fører ofte til problemer for bioinformatikere som vil bruke databaser som grunnlag for videre analyser, men også til forvirring blant ”vanlige” biologer som vil studere studere strukturen. Eksempelproblem: proteiner med lederpeptider
: sekvensen av hele proteinet. Implisitt sekvens: sekvensen som ligger i strukturfilen. Siden strukturfiler ofte mangler noen residuer (f eks på N-terminus) er implisitt og eksplisitt sekvens ofte ikke like. Dette fører ofte til problemer for bioinformatikere som vil bruke databaser som grunnlag for videre analyser, men også til forvirring blant vanlige biologer som vil studere studere strukturen. Eksempelproblem: proteiner med lederpeptider.")
53
Kitinbindende protein fra Serratia marcescens; sekretert etter spalting av et leder peptid på mellom 22 og 45 aminosyrer. Hvilke residunummer skal man bruke når man sammenligner disse sekvenser og tilhørende strukturer?

54
Datafiler med strukturinformasjon
PDB-format Liste over atomkoordinater Programvaren (programmereren!) tolker avstandene ut i fra liste over bindingsavstander (eks C-C er ca 1,5 Å; ”chemistry rules approach”) Ingen fastlagt måte å tolke unntak på Bare eksplisitt sekvens MMDB-format PDB-derivat som egner seg for rask databehandling Implisitt sekvens Eksplisitt bonding approach
 tolker avstandene ut i fra liste over bindingsavstander (eks C-C er ca 1,5 Å; chemistry rules approach ) Ingen fastlagt måte å tolke unntak på. Bare eksplisitt sekvens. MMDB-format. PDB-derivat som egner seg for rask databehandling. Implisitt sekvens. Eksplisitt bonding approach.")
55
Entrez structure Tilgjengelige databaser har massevis av nyttige muligheter, for eksempel BLAST mot alle PDB sekvenser og ”derived” databaser med ”sequence neighbours” av proteiner med kjent struktur

56
Nøyaktighet i krystalstrukturer
B-faktorer: Sier noe om hvor bra atomet passet i elektrontettheten; jo høyere B-faktor, jo mer usikker og det skyldes beveglighet og/eller unnøyaktighet. Det er normal at B-faktorene varierer og av og til er høye (f eks overflate residuer), men høye B-faktorer i en hel region kan tyde på feil i strukturbestemmelse. Resolution: sier noe om hvor detaljert informasjonen er (høyere resolusjon betyr mer observasjoner per parameter). Jo mer orden i krystallen, jo bedre resolusjon og jo mer man kan ”se” i strukturen. Alt under 2.5 – 3.0 Å er bra, mens man under 2 Å vanligvis kaller resolusjonen ”høy” To type feil i tillegg til det som bestemmes av selve krystallen: data innsamling og strukturløsning. Feielen kan bli vesentlige (opp til 0.5 Å mellom strukturer som burde være ”identiske” – se også Fig. 5.8a, s. 235). Det finnes kontroleprogrammer som automatisk rapporterer ”stereochemical outliers”, som igjen kan tyde på feil (men disse er ikke nødvendigvis virkelige feil!) a) atom number: atoms are numbered in sequence through the file; b) atom type: n = amide N, ca = alpha C, c = carbonyl C, o = carbonyl O, cb = beta carbon, and so forth; c) residue name: three-letter amino acid abbreviation; d) residue number; e) x-coordinate of the atom, in angstroms from the unit-cell origin; f) y-coordinate of the atom; g) z-coordinate of the atom; h) occupancy: the fraction of unit cells that contain the atom in this particular location, usually 1.00, or all of them (can be used to represent alternative conformations of side chains); i) temperature factor: an indication of uncertainty in this atom's position due to disorder or thermal vibrations (can be used by graphics programs to represent the relative mobility of different parts of a protein) j) every line ends with the PDB file identification code.
, men høye B-faktorer i en hel region kan tyde på feil i strukturbestemmelse. Resolution: sier noe om hvor detaljert informasjonen er (høyere resolusjon betyr mer observasjoner per parameter). Jo mer orden i krystallen, jo bedre resolusjon og jo mer man kan se i strukturen. Alt under 2.5 – 3.0 Å er bra, mens man under 2 Å vanligvis kaller resolusjonen høy To type feil i tillegg til det som bestemmes av selve krystallen: data innsamling og strukturløsning. Feielen kan bli vesentlige (opp til 0.5 Å mellom strukturer som burde være identiske – se også Fig. 5.8a, s. 235). Det finnes kontroleprogrammer som automatisk rapporterer stereochemical outliers , som igjen kan tyde på feil (men disse er ikke nødvendigvis virkelige feil!) a) atom number: atoms are numbered in sequence through the file; b) atom type: n = amide N, ca = alpha C, c = carbonyl C, o = carbonyl O, cb = beta carbon, and so forth; c) residue name: three-letter amino acid abbreviation; d) residue number; e) x-coordinate of the atom, in angstroms from the unit-cell origin; f) y-coordinate of the atom; g) z-coordinate of the atom; h) occupancy: the fraction of unit cells that contain the atom in this particular location, usually 1.00, or all of them (can be used to represent alternative conformations of side chains); i) temperature factor: an indication of uncertainty in this atom s position due to disorder or thermal vibrations (can be used by graphics programs to represent the relative mobility of different parts of a protein) j) every line ends with the PDB file identification code.")
57
Mulige mangler i krystalstrukturer
Mange småfeil er mulig (disse finner man med ”check-programmer”; eksempel RW Hooft et al., Nature 381:272, 1996; Manglende data pga fleksibilitet Manglende data pga at man bare har krystallisert et protein fragment Krystalkontakter Molekylet er ”frossen”; dynamikken er skjult Husk også: Det finnes ”ambiguity” mht tilordning av sekundær struktur Erfaringsdatabasen består stort sett av globulære proteiner Krystallografi ekstremt nyttig og meget presiss. De fleste strukturer representer nok mer eller mindre ”sannheten”

58
Strukturer bestemt med NMR
Struktur i løsning Svaret av NMR studiet består av en ”ensemble” med strukturer som er kompatible med de eksperimentelle data. Dette viser reelle bevegelser. Mindre nøyaktig enn krystallografi og dermed mindre egnet for studier av strukturelle detaljer. Svært verdifull teknikk fordi den kan en del som ikke X-ray krystallografi kan (dynamikk, interaksjoner) Mindre ”vanlig” teknikk enn krystallografi (dvs fære strukturer i PDB)
 Mindre vanlig teknikk enn krystallografi (dvs fære strukturer i PDB)")
59
Programmer til visualisering
RasMol (RasWin/RasMac)/Chime Visualisering av kjent 3D struktur Åpner datafilene, men kan ikke redigere dem, men man kan lage scripts som viser modellen på en bestemt måte Network entrez - Cn3D Visualisering av kjent 3D struktur og strukturelle “naboer” Cn3D kan lagre bestemte visualiseringer av modellene SwissPDBviewer Visualisering, modellering, threading mm. Meget kraftig, kan også eksportere modeller for ekstern ”rendering” med PovRay. Protein Explorer se
/Chime. Visualisering av kjent 3D struktur. Åpner datafilene, men kan ikke redigere dem, men man kan lage scripts som viser modellen på en bestemt måte. Network entrez - Cn3D. Visualisering av kjent 3D struktur og strukturelle naboer Cn3D kan lagre bestemte visualiseringer av modellene. SwissPDBviewer. Visualisering, modellering, threading mm. Meget kraftig, kan også eksportere modeller for ekstern rendering med PovRay. Protein Explorer. se")
60
Visualisering av strukturer – mange muligheter
Cartoon Alle atomer Bovint papillomavirus DNA-bindende protein E2, visualisert med RasMol Dataprogrammet har tilordner sekundær struktur-elementer Bildekvalitet er sterkt avhengig av programmet; Rasmol er ikke spesielt bra

61
Webviewer fra Molecular Simulations
Visualisering Webviewer fra Molecular Simulations

62
Avansert visualisering fra kitinase forskningen
Asp215 Gln144 Tyr10 Asp142

63
Avansert visualisering
Disse bilder: Pymol Legge merke til at programmer kan være laget for visualisering, modellering/analyser osv, eller for begge. Er man bare ute etter fine bilder bør man bruke et visualiseringsprogram og ikke et modelleringsprogram som f eks WHAT IF

64
Avansert visualisering

65
Avansert visualisering Interpolering mellom krystalstrukturer kan gi en film

66
Visualisering – hva finner du i struktur filen?
Crystallographic Unit: hvis den inneholder flere enn 1 proteinmolekyl, vil programmene lese inn alle, mens du kanskje bare vil se på en (Nb. Dette kan altså også forekomme for ikke multimere proteiner) NMR strukturfiler inneholder som oftest en ”ensemble” av strukturer Farget etter B-faktor NMR struktur interleukin 4; 1bcn.pdb
 NMR strukturfiler inneholder som oftest en ensemble av. strukturer. Farget etter B-faktor. NMR struktur interleukin 4; 1bcn.pdb.")
67
PROTEIN STRUKTUR: Konservering Sammenligning Klassifisering

68
Struktur er bedre konservert enn sekvens
Protein ”structure space” er begrenset; naturen bruker en begrenset (1000?) antall strukturer (”folds”), og mange proteiner har omtrent samme struktur, selv uten detekterbar sekvenslikhet All strukturer består av sekundær struktur elementer pluss ”loops” og ”coil”; proteiner har en hydrofob kjerne Det finnes predikerbare forhold mellom sekvenslikheter og struktur, f eks: Residuer i den hydrofobe kjernen viser en annen ”substitusjonsmønster” enn residuer utenfor (derfor bruker man gjerne ”scoringsmatrikser” ved sammenligning av proteinsekvenser) Insersjoner og delesjoner forekommer som oftest utenfor kjernen, i ”loop” eller ”coil” regioner Proteiner som er homologe har samme struktur; proteiner som viser strukturlikhet (ofte kallet ”strukturell homologi”) er sannsynligvis homologe (sekvens har evoluert mer enn struktur)
 antall strukturer ( folds ), og mange proteiner har omtrent samme struktur, selv uten detekterbar sekvenslikhet. All strukturer består av sekundær struktur elementer pluss loops og coil ; proteiner har en hydrofob kjerne. Det finnes predikerbare forhold mellom sekvenslikheter og struktur, f eks: Residuer i den hydrofobe kjernen viser en annen substitusjonsmønster enn residuer utenfor (derfor bruker man gjerne scoringsmatrikser ved sammenligning av proteinsekvenser) Insersjoner og delesjoner forekommer som oftest utenfor kjernen, i loop eller coil regioner. Proteiner som er homologe har samme struktur; proteiner som viser strukturlikhet (ofte kallet strukturell homologi ) er sannsynligvis homologe (sekvens har evoluert mer enn struktur)")
69
Struktur og sekvens likheter
Proteiner med > 25 % sekvenslikhet har lignende strukturer og er homologe Derfor er predikjson av protein strukturer ( i hvert fall i grove trekk) i prinsipp mulig for en rekke proteiner Se også side og figur 5.8A
 i prinsipp mulig for en rekke proteiner. Se også side og figur 5.8A.")
70
Men……. Proteiner med < 25 % sekvenslikhet eller ingen detekterbar sekvenslikhet i det hele tatt KAN ha lignende strukturer og KAN være homologe Sammenligning av strukturer kan derfor gi informasjon om likheter som sammenligning av sekvenser ikke gir

71
Struktur er bedre konservert enn sekvens
Proteiner med > 25 % sekvenslikhet har lignende strukturer og er homologe; disse proteiner tilhører samme ”familie”. Hva betyr ”lignende strukur”, ”samme struktur”, ”samme fold”, osv ? Lignende ”overall” organisering av minst en del av proteinet Jo lavre sekvenslikhet, jo større de strukturelle forskjellene ”Lignende” er til dels et definisjonsspørsmål; forskjellige grupper kommer til forskjellige konklusjoner (ved lav likhet) Legg merke til at en forskjell på f eks 0.5 Å i en side kjede mellom de aktive setene av to enzymer kan ha stor funksjonell betydning, mens enzymene uten tvil vil ble bedømt til å ha ”lignende struktur”
 Legg merke til at en forskjell på f eks 0.5 Å i en side kjede mellom de aktive setene av to enzymer kan ha stor funksjonell betydning, mens enzymene uten tvil vil ble bedømt til å ha lignende struktur")
72
Protein struktur, terminologi
Arkitektur: Arrangementet av SSEer i forhold til hverandre Topologi: Hvordan disse SSE er forbundet med hverandre Fold: Arrangement av SSEer som er topologisk definert; ”Motif” og ”supersekundærstruktur” er lignende begrep men brukes som oftest for mindre store enheter av struktur (jfr f eks ”helix-turn-helix” motif) Family: Proteiner med sekvenslikhet (> X %; X = 25 – 50 %) [begrepet brukes også i strukturell kontekst for proteiner som har strukturlikhet] Superfamily: Gruppe familier som ser ut til å være evolusjonært beslektet (lite sekvenslikhet, men klare strukturelle og/eller funksjonelle likheter) Class: ”groveste” inndeling av proteiner: f eks. a, b, a+b, a/b
 Family: Proteiner med sekvenslikhet (> X %; X = 25 – 50 %) [begrepet brukes også i strukturell kontekst for proteiner som har strukturlikhet] Superfamily: Gruppe familier som ser ut til å være evolusjonært beslektet (lite sekvenslikhet, men klare strukturelle og/eller funksjonelle likheter) Class: groveste inndeling av proteiner: f eks. a, b, a+b, a/b.")
73
Protein struktur, Terminologi
Domene: Kan sees i en strukturelt og/eller en sekvens kontekst. Sekvens: to-domene protein A kan ha en del som ligner på protein B og en del som ligner på protein C -> to sekvensdomener Strukturelt: (mer eller mindre) “uavhengige” deler av proteinet, ofte med eget hydrofobt indre; man vet eller antar ofte at domener også er uavhengige ”foldons”.
 uavhengige deler av proteinet, ofte med eget hydrofobt indre; man vet eller antar ofte at domener også er uavhengige foldons .")
74
Hva er et “domene” Et segment som har knyttet til seg funksjon?
Kjernelokalisering (NLS), DNA-binding osv. Ofte gjenkjent i endimensjonal sekvenssøk Et segment som vi ser konservert under evolusjon? Exon shufling etc. En del av proteinet med separat hydrofob kjerne mm. Strukturkunnskap nødvendig En del av proteinet som kan folde uavhengig (foldon) Et strukturelt domene behøver ikke bestå av et sammenhengende stykke peptidkjede Domener kan foreslås av algoritmer, men bør vurderes av eksperter OBS! Domenbegrepet brukes på mange forskjellige måter; ikke alt som står her gjelder for hver domen!
, DNA-binding osv. Ofte gjenkjent i endimensjonal sekvenssøk. Et segment som vi ser konservert under evolusjon Exon shufling etc. En del av proteinet med separat hydrofob kjerne mm. Strukturkunnskap nødvendig. En del av proteinet som kan folde uavhengig (foldon) Et strukturelt domene behøver ikke bestå av et sammenhengende stykke peptidkjede. Domener kan foreslås av algoritmer, men bør vurderes av eksperter. OBS! Domenbegrepet brukes på mange forskjellige måter; ikke alt. som står her gjelder for hver domen!")
75
Modulære proteiner

76
Domener i thermolysin Thermolysin:
Det aktive sete ligger mellom to ”domener” Det C-terminale domene kan videre deles opp i to subdomener Man vet at den gule delen folder uavhengig

77
Domener versus motiver versus subenheter
Motif: et karakteristisk struktur eller sekvens element; ikke nødvendigvis en domene Subenheter: individuelle proteiner som assosierer til å danne komplekser; det finnes proteiner som er multimere i en organisme og multidomene (monomere) i en annen.
 i en annen.")
78
Ca distance plotting for å finne domener
Ca - Ca distance plot of g-crystallin Større distansen gir mer svart Dette ser ut til å være et to-domen protein Residu no Residu no

79
Sammenligning og klassifisering av proteinstrukturer
Strukturene klassifiseres på forskjellige måter og med varierende menneskelig medvirkning ut fra SSE-innhold, arkitektur, topologi, fold, class, family, superfamily mm. Dette gjør man fordi man vil: Få oversikt over naturen (antall ”folds”) Oppdage funksjonelle og evolusjonære relasjoner mellom proteiner Man bruker vanligvis domener Mht klassifisering finnes ingen ”sannhet”; Mange grensetilfeller Terminologien er ikke helt konsistent
 Oppdage funksjonelle og evolusjonære relasjoner mellom proteiner. Man bruker vanligvis domener. Mht klassifisering finnes ingen sannhet ; Mange grensetilfeller. Terminologien er ikke helt konsistent.")
80
Enkel klassifisering av protein strukturer (classes)
Det finnes flere måter å gjøre dette på, f eks: Bare a Bare b a/b: blandet alpha-heliks og beta-strands a + b: alpha-heliks del mer eller mindre separert fra beta-del Multidomen proteiner (dvs flere ”classes”) Membranproteiner (Se også side 43 – 48)
 Membranproteiner. (Se også side 43 – 48)")
81
Avansert sammenligning av protein-strukturer (s. 230-237)
Dette gir mer informasjon enn sekvensalignment og det kan føre til nye oppdagelser fordi struktur er bedre konservert enn sekvens. Eksempel: konserverte aktive seter. Dette synliggjør evolusjonære relasjoner på en måte som sekvens alignments ikke kan (ved lav sekvens likhet) Man får en sekvens alignment som gir ”sannheten” (OBS! et meget viktig poeng!) Man får et utgangspunkt for klassifisering av proteiner (domener) etter struktur
 Man får en sekvens alignment som gir sannheten (OBS! et meget viktig poeng!) Man får et utgangspunkt for klassifisering av proteiner (domener) etter struktur.")
82
Sammenligning av proteinstruktur
Det er ikke lett å sammenligne in computo fordi man trenger å filtrere bort likheter uten mening (sekundære struktur elementer). Man må bruke triks for å få akseptabel CPU bruk. Viktig triks: man beskriver proteiner i første omgang som en samling ordnede SSE’s. To overlappende SSE’s danner en ”unit of structural similarity”. Programmer (eksempler): VAST (SSE-basert; Vector-Alignment Search Tool); database: MMDB – Entrez DALI (basert på kontakt-nettverk; Distance Matrix Alignment Program); database: FSSP (Families of structurally similar proteins). OBS! Mye brukt. (side 232 i boken)
. Man må bruke triks for å få akseptabel CPU bruk. Viktig triks: man beskriver proteiner i første omgang som en samling ordnede SSE’s. To overlappende SSE’s danner en unit of structural similarity . Programmer (eksempler): VAST (SSE-basert; Vector-Alignment Search Tool); database: MMDB – Entrez. DALI (basert på kontakt-nettverk; Distance Matrix Alignment Program); database: FSSP (Families of structurally similar proteins). OBS! Mye brukt. (side 232 i boken)")
83
SARF-algoritmen (Spatial ARangement of backbone Fragments):
1. Tilordne SSEer for hvert protein 2. Søke etter kompatible par av SSEer i de to proteinene 3. Søke etter større strukturlikheter forankret i de kompatible SSE-parene 4. Finjustering av strukturtilpasningen

84
SARF-algoritmen, SSE tilordning:
Sammenligner (kun) Ca-koordinater fra to og to PDB-filer Dynamisk programmering: sammenligner vindu på 5 Ca-er med prototype a-heliks og b-strand, og teller som positivt hvis rmsd<hhv. 0,4 eller 0,8Å. [man tar altså ikke hensyn til H-bindinger. Dette kan gi avvik spes. for -strand] Hvert SSE beskrives av en vektor
 Ca-koordinater fra to og to PDB-filer. Dynamisk programmering: sammenligner vindu på 5 Ca-er med prototype a-heliks og b-strand, og teller som positivt hvis rmsd<hhv. 0,4 eller 0,8Å. [man tar altså ikke hensyn til H-bindinger. Dette. kan gi avvik spes. for -strand] Hvert SSE beskrives av en vektor.")
85
SARF, påvisning av største SSE ensemble:
For å unngå ”kombinatorisk eksplosjon” utelukkes SSEer som ligger >25Å fra ensemblet fjerner også ”støy” Programmet går gjennom opptil flere tusen ensembler per proteinpar Optimalisering delesjoner i SSEene og søker etter ytterligere SSEer Iterativ (4 –5 runder) Mot minimal rmsd
 Mot minimal rmsd.")
86
DALI (side 232) Mye brukt Basert på observasjonen at kontakter pleier å være konservert (f eks: når to residuer begge blir litt mindre under evolusjonen, så endrer strukturen seg ofte på en slik måte at disse to residuer kommer nærmere -> kontakten beholdes) DALI er basert på deteksjon av konserverte kontakt-mønstre. Mha DALI har mange nye strukturelle likheter blitt oppdaget (og dermed mulige evolusjonære slektskap)
 DALI er basert på deteksjon av konserverte kontakt-mønstre. Mha DALI har mange nye strukturelle likheter blitt oppdaget (og dermed mulige evolusjonære slektskap)")
87
Klassifisering av strukturer
Det finnes mange databaser (f eks FSSP) hvor alle tilgjengelige protein strukturer er delt opp i ”strukturelle familier”, dvs i grupper av proteiner med strukturell likhet. Denne inndelingen kan være basert på strukturalignments (f eks DALI for FSSP), eller på en type kategorisering (dvs veldig forskjellige typer inndeling) Det er som oftest nyttig å prøve flere databaser hvis du leter etter noe som ikke er ”obvious” Strukturdatabaser gir links til disse ”classification databases
 hvor alle tilgjengelige protein strukturer er delt opp i strukturelle familier , dvs i grupper av proteiner med strukturell likhet. Denne inndelingen kan være basert på strukturalignments (f eks DALI for FSSP), eller på en type kategorisering (dvs veldig forskjellige typer inndeling) Det er som oftest nyttig å prøve flere databaser hvis du leter etter noe som ikke er obvious Strukturdatabaser gir links til disse classification databases.")
88
Strukturlikhet Strukturelle “naboer” via Entrez
Hele MMDB er på forhånd sammenlignet med seg selv Med VAST-algoritme Man kan få frem en liste Med strukturelle naboer Vha. hjelpeprogrammet Cn3D kan disse visualiseres sammen

89
PDB - Structural neighbours

90
Klassifisering FSSP: strukturelle likheter fra struktursammen-ligninger med DALI (”Fold classification based on Structure-Structure alignment of Proteins”); også kalt DDD: ”Dali Domain Dictionary”) CE: database av strukturalignments CATH: ”Class, Architecture, Topology, Homology” SCOP: ”Structural Classification Of Proteins” (side )
; også kalt DDD: Dali Domain Dictionary ) CE: database av strukturalignments. CATH: Class, Architecture, Topology, Homology SCOP: Structural Classification Of Proteins (side )")
91
Hierarkisk klassifisering; http://scop.mrc-lmb.cam.ac.uk/scop/
Family: likhet i struktur, sekvens, og, av og til, funksjon viser evolusjonær slektskap Superfamily: samling av familier hvor det kan se ut som om det finnes slektskap mellom familiene

92
Alignment av strukturer, noen viktige poeng til slutt
RMSD: Root mean square deviation i Å: sier noe om hvor like strukturene er. Rmsd ligger under 0.5 Å for ”identiske” strukturer. OBS! Legg merke til at en strukturell likhet mellom to proteiner som har mer en 25 % sekvens likhet er ”obvious”. Derfor jobber f eks FSSP med en subsett av ”representativ” eller ”non-redundant” structures” (dvs at man ikke tar med to strukturer av proteiner med > 25 % sekvens likhet) En strukturbasert sekvens alignment er den mest pålitelige type sekvens alignment som finnes
 En strukturbasert sekvens alignment er den mest pålitelige type sekvens alignment som finnes.")
93
Alignment av strukturer, noen viktige poeng til slutt
Det finnes også HSSP (”homology-derived structures of proteins”); HSSP er en database av strukturell signifikante sekvens alignments, hvor minst et av sekvensen i hver alignment representerer et protein med kjent struktur; dette er, mao, en oversikt over ”obvious” implisitt eller eksplisitt strukturell likhet DALI/FSSP er også en database av eksplisitte multiple strukturbaserte sekvensalignements ”in the twilight zone of sequence similarity”. For mer info se f eks Nucleic Acids Research 26: (1998)
; HSSP er en database av strukturell signifikante sekvens. alignments, hvor minst et av sekvensen i hver alignment representerer et. protein med kjent struktur; dette er, mao, en oversikt over obvious implisitt eller eksplisitt strukturell likhet. DALI/FSSP er også en database av eksplisitte multiple strukturbaserte. sekvensalignements in the twilight zone of sequence similarity . For mer. info se f eks Nucleic Acids Research 26: (1998)")
94
MULTIPLE SEKVENS ALIGNMENTS (MSA) PROFILER
 PROFILER")
95
Multiple sekvens alignments
MSAs gir mye mer informasjon en enkle alignments; man kan f eks finne ”sekvens profiler” som har betydning: ARLEVSANFT ARLEVSANFT LQVKINLDLK LQVKINLDLK MEANATIQAQ HPHPHPHPHP MSA gir en ”trygghet” som gjør at man kan få ut flere konklusjoner MSAs er startpunkt for nesten alle moderne metoder for prediksjon av struktur og funksjon på basis av sekvens (som vi vil behandle senere)
")
96
MSAs gir strukturell informasjon
MSAs gir et godt utgangspunkt fordi de synliggjør mønstre som tyder på bestemte typer sekundær struktur: Korte hydrofobe strekninger: buried b-strand i, i + 2, i + 4 mønstre med hydrofobe residuer: b-strand som ligger på overflaten i, i + 3, i + 4, i + 7; mønstre: a-heliks insersjoner og delesjoner har en preferanse for ”surface loops” Konserverte glyciner og proliner indikerer spesielle typer strukturelementer (”loops” eller ”turns”) Sterkt konserverte områder kan tyde på f eks en aktive sete
 Sterkt konserverte områder kan tyde på f eks en aktive sete.")
97
Om MSA metoder: En struktur-basert alignment er per definisjon best
Software for sekvens alignment blir derfor testet med test-setts med sekvenser av proteiner med kjent struktur. Mao: en perfekt multiple sekvens alignment viser hvilke residuer har samme posisjon i den tre-dimensjonale strukturen. Det å lage en MSA for sekvenser som ikke er spesielt like er langt fra trivielt, selv om man bruker tilgjengelige, profesjonelle programmer som ClustalW. Det finnes databaser med sekvens alignments (f eks HSSP). Disse bør man bruke hvis det er mulig.
. Disse bør man bruke hvis det er mulig.")
98
Framgangsmåten -> Dette er en iterativ, delvis manuell prosess!
Finn sekvensene Edit sekvenser slik at de blir omtrent like lange (dvs ta bort ekstra domener osv som helt klart ikke forekommer i de andre proteinene) Kjør noen første forsøk, f eks ”pairwise” for å optimalisere dette Kjør MSA program (f eks Clustal W) Se på resultatet; editer sekvenser; tilfør eller slett sekvenser, bruk struktur-informasjon hvis tilgjengelig Til slutt, når du har en tilsynelatende bra alignment, prøv å tilføre de mer ”vanskelige” sekvenser -> Dette er en iterativ, delvis manuell prosess!
 Kjør noen første forsøk, f eks pairwise for å optimalisere dette. Kjør MSA program (f eks Clustal W) Se på resultatet; editer sekvenser; tilfør eller slett sekvenser, bruk struktur-informasjon hvis tilgjengelig. Til slutt, når du har en tilsynelatende bra alignment, prøv å tilføre de mer vanskelige sekvenser. -> Dette er en iterativ, delvis manuell prosess!")
99
Problemer Problemer forekommer først og fremst i nærheten av insersjoner og delesjoner: Når man tviler om man har signifikant likhet kan man utføre en Monte-Carlo test of significance (randomiser sekvensen, med behold av lengde og aminosyresammensetning og align; gjør det f eks 100 ganger)
")
100
Hierarkiske metoder Pairwise aligments Guided tree Alignment
Variasjoner: Scoringsmatrikser Håndtering av ”feil” som oppstår tidlig i prosessen Gap opening og extension penalties Variasjon i gap penalties ( f eks ingen gaps tillatt i det som er eller ser ut til å være sekundær struktur elementer)
")
101
Hierarchical methods

102
Clustal W Hierarkisk metode Lett å bruke; mange web-services
Secondary structure masks (bias gap posisjonering) Man kan bruke alignments som input Bør kombineres med et program til visualisering [Nb. Det finnes andre programmer……]
 Man kan bruke alignments som input. Bør kombineres med et program til visualisering. [Nb. Det finnes andre programmer……]")
103
Analyse av MSA’s

104
Analyse av MSA’s Det finnes mange programmer for visualisering og ”highlighting”, f eks ALSCRIPT: Identisk i alle sekvenser: hvit på grå, og i bokser. Similaritet, dvs like kjemiske egenskaper: svart på lysegrå og i bokser. Sekundær struktur elementer er indikert OBS! Husk at det ikke er trivielt å si hvilke residuer er like eller ikke; det er delvis avhengig av i hvilken kontekst man analyser en MSA.

105
Bruk av MSAs for protein identifikasjon
Profiler PSI-BLAST Hidden Markov Models (HMMs)
")
106
Profiler (s. 190 – 192, Plate VII)
Man lager en ”inventory array” for en MSA som man bruker sammen med en substitusjonsmatriks til å lage en ”position specific scoring matrix” (som inkluderer gaps) Har man en ukjent sekvens, så kan man søke med den mot en database av slike profiler En aminosyre får høy score når (1) den forekommer ofte i en posisjon, eller (2) når den har en høy sannsynlighet å erstatte det mer vanlige residuen på denne posisjonen under evolusjonen Det finnes grafiske metoder for å vise en profil (Plate VIIB)
 Har man en ukjent sekvens, så kan man søke med den mot en database av slike profiler. En aminosyre får høy score når (1) den forekommer ofte i en posisjon, eller (2) når den har en høy sannsynlighet å erstatte det mer vanlige residuen på denne posisjonen under evolusjonen. Det finnes grafiske metoder for å vise en profil (Plate VIIB)")
107
PSI-BLAST Position-specific iterated BLAST: dvs at initielle hits blir brukt til å lage en MSA som så bli brukt til å lage en profil -> nytt søk -> forbedret profil -> nytt søk I området med < 30 % sekvenslikhet finner PSI-BLAST tre ganger mer signifikante hits enn BLAST; nyttig for genomannotering Det eneste som er bedre enn PSI-BLAST er HMM-baserte metoder eller metoder hvor strukturell informasjon tas med eksplisitt

108
HMMs Nyttig statistisk metode for å analysere og utnytte subtile sekvensmønstre i MSAs Ny sekvenser kan analyseres mot en sett av HMMs som representerer en sett med MSAs (F eks i Pfam) Se side for flere detaljer
 Se side for flere detaljer.")
109
PRESISERING Profile: ARLEVSANFT LQVKINLDLK HPHPHPHPHP
MEANATIQAQ HPHPHPHPHP Motif: konservert sekvens mønster; kan være veldig kort, f eks 3 – 10 aminosyrer rundt en aktive sete, en glykosileringssete osv. Database: Prosite, verktøy: Scan Prosite. Her brukes også begrepene ”pattern” og ”signature”. Disse kan for eksempel se slik ut: [IV] – G – x – G – T – [LIVMF] – x(2) – [GS] MSA
 – [GS] MSA.")
110
Sekvens versus struktur alignments
Teknisk sett er alignment av sekvenser og alignment (superponering) av strukturer to forskjellige ting. MEN: det ligger veldig mye ”struktur” i en alignment og en multiple sekvens alignment (og profilene som følger derfra) gir meget nyttig informasjon for videre strukturelle analyser og prediksjoner. Eksempel: bygging av tre-dimensjonale modeller starter med en alignment, som i praksis sier det meste om hvordan modellen kommer til å se ut i grove trekk (se nedenfor). Eksempel: mange av prediksjonsmetodene som behandles nedenfor
 av strukturer to forskjellige ting. MEN: det ligger veldig mye struktur i en alignment og en multiple sekvens alignment (og profilene som følger derfra) gir meget nyttig informasjon for videre strukturelle analyser og prediksjoner. Eksempel: bygging av tre-dimensjonale modeller starter med en alignment, som i praksis sier det meste om hvordan modellen kommer til å se ut i grove trekk (se nedenfor). Eksempel: mange av prediksjonsmetodene som behandles nedenfor.")
111
STRUKTUR PREDIKSJON DEL 1: Sekvens-baserte prediksjoner av funksjon og diverse detaljer

112
Prediksjoner på basis av sekvens
Vi kjenner mange gener og proteiner, men vi vet ikke like mye om hvert protein. Stor gap mellom antallet kjente sekvens og antallet kjente strukturer. Enorm variasjon i strukturer, kjemiske egenskaper og funksjonalitet Prediksjon av egenskaper på basis av bare sekvens er nyttig. Sentral dogma: sekvens bestemmer struktur (og det faktum at det finnes chaperoner endrer ikke på dette)
")
113
www.expasy.ch Å bestemme egenskaper til kjente proteiner
Å identifisere ukjente proteiner på basis av egenskapene Strukturmodel-lering og visualisering Og mye, mye mer………

117
OBS! Kan være ulik virkelig pI

118
Expasy; metoder for identifikasjon
AACompident & AACompSim: Søke etter likheter mellom proteiner ved å sammenligne aminosyre- sammensetning eventuelt kombinert med andre ting man kan finne ut om proteinet (pI, MW, taxonomic class) -> å identifisere ukjente proteiner og (i noen få tilfeller) å finne ukjente homologer av et kjent protein. PROPSEARCH Samme som AACompSIM, men man bruker 144 egenskaper (f eks gjennom- snittlig hydrofobisitet, innhold med ”bulky” residuer, osv) MOWSE ( Molecular Weight Search algorithm: protein identifkasjon på basis av molecular weight (fra Masse spektrometri) for hele proteinet, eller for fragmenter, generert Med spesifikke proteaser (som trypsin- som kutter bak Lys og Arg). Man søker mot en database av ”non-redundant” protein sekvenser (OWL) -> viktig verktøy i proteomics OBS! Det skjer veldig mye innen proteomics (dvs utviklinger a la MOWSE) OBS! Eksperimentelle aminosyresammensetninger er ofte unøyaktige
 -> å identifisere ukjente proteiner og (i. noen få tilfeller) å finne ukjente homologer av et kjent protein. PROPSEARCH. Samme som AACompSIM, men man bruker 144 egenskaper (f eks gjennom- snittlig hydrofobisitet, innhold med bulky residuer, osv) MOWSE ( Molecular Weight Search algorithm: protein identifkasjon på basis av molecular. weight (fra Masse spektrometri) for hele proteinet, eller for fragmenter, generert. Med spesifikke proteaser (som trypsin- som kutter bak Lys og Arg). Man søker. mot en database av non-redundant protein sekvenser (OWL) -> viktig verktøy i proteomics. OBS! Det skjer veldig mye innen proteomics (dvs utviklinger a la MOWSE) OBS! Eksperimentelle aminosyresammensetninger er ofte unøyaktige.")
119
Signal P: kjenner igjen signalsekvenser; viktig ved f eks genom
annotering og ”genome mining”

120
Sekvensbasert prediksjon av funksjon; MSAs
Overall sekvens likhet – proteinfamilier; persentasje likhet innad en familie varierer fra 25 % til 50 % (i PIR, Protein Information Resource); inkluderer FSSP/HSSP Moduler: kortere områder med sekvens likhet, f eks i proteiner som bare er like i en del av sekvensen; begrepet brukes blant annet for å se etter superfamilier Tilstedeværelse av bestemte mønstre / motif av varierende størelse og karakter. Eksempel: Prosite databasen -> aktive seter, glykosileringsseter osv osv
; inkluderer FSSP/HSSP. Moduler: kortere områder med sekvens likhet, f eks i proteiner som bare er like i en del av sekvensen; begrepet brukes blant annet for å se etter superfamilier. Tilstedeværelse av bestemte mønstre / motif av varierende størelse og karakter. Eksempel: Prosite databasen -> aktive seter, glykosileringsseter osv osv.")
121
Sekvensbasert prediksjon
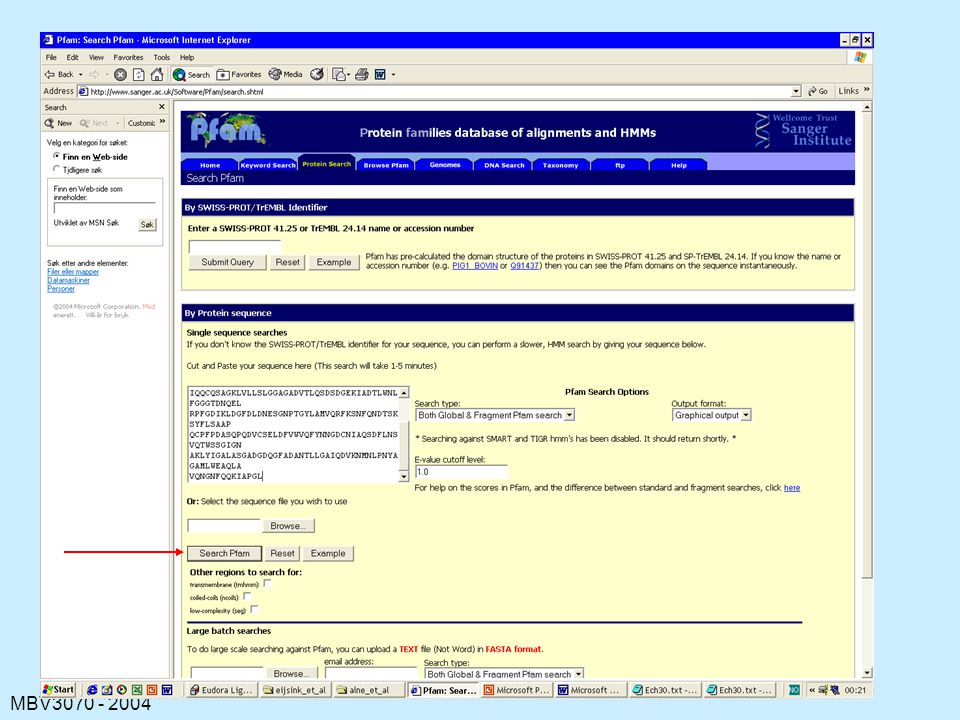
Det finnes massevis av databaser (og tilhørende søkemotorer for å analysere ukjente sekvenser) med varierende typer sekvens- basert eller sekvens-orientert informasjon. Vi fokuserer på: BLAST, PSI-BLAST (se ovenfor) BLOCKS Pfam (svært viktig) Prosite INTERPRO: flere databasesøk samtidig -> svært viktig
 med varierende typer sekvens- basert eller sekvens-orientert informasjon. Vi fokuserer på: BLAST, PSI-BLAST (se ovenfor) BLOCKS. Pfam (svært viktig) Prosite. INTERPRO: flere databasesøk samtidig -> svært viktig.")
122
Blocks Block: ”stor motif” eller kombinasjon av motiver som
Kilde-motif Blocks Block: ”stor motif” eller kombinasjon av motiver som ligger etter hverandre og som kan bli aligned uten gap Søk: din sekvens blir aligned mot alle Blocks i alle mulige posisjoner og man beregner en score Sekvens- likhet Nummer på første aminosyre (fra oppr.sekvens) Swiss-Prot Accession number Sekvenser er gruppert etter sekvenslikhet
 Swiss-Prot. Accession. number. Sekvenser er gruppert etter sekvenslikhet.")
123
Blocks

124
Blocks

125
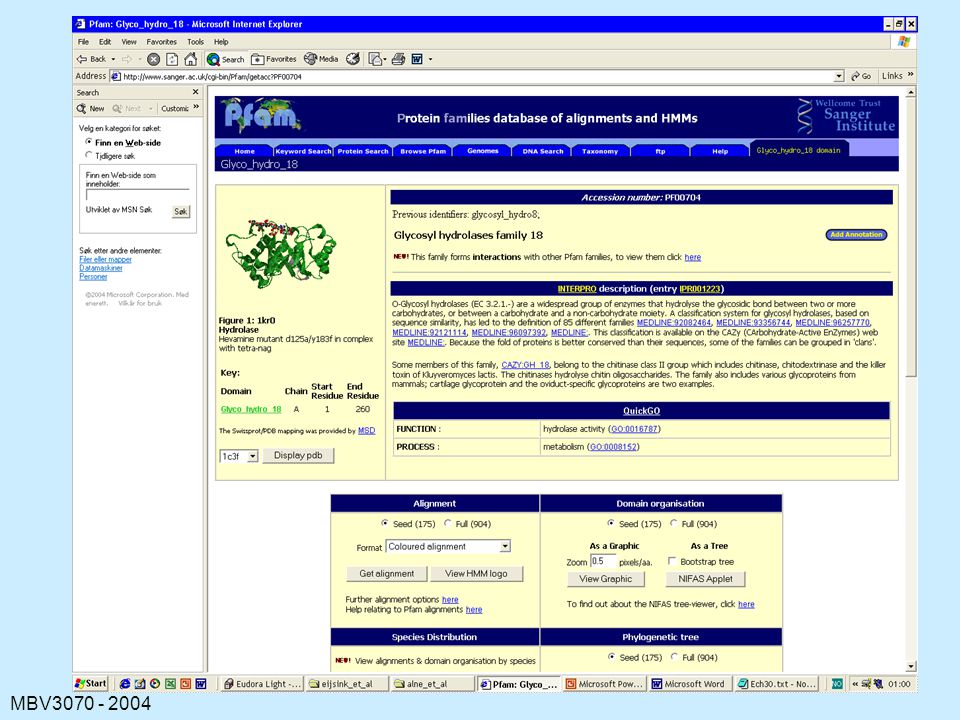
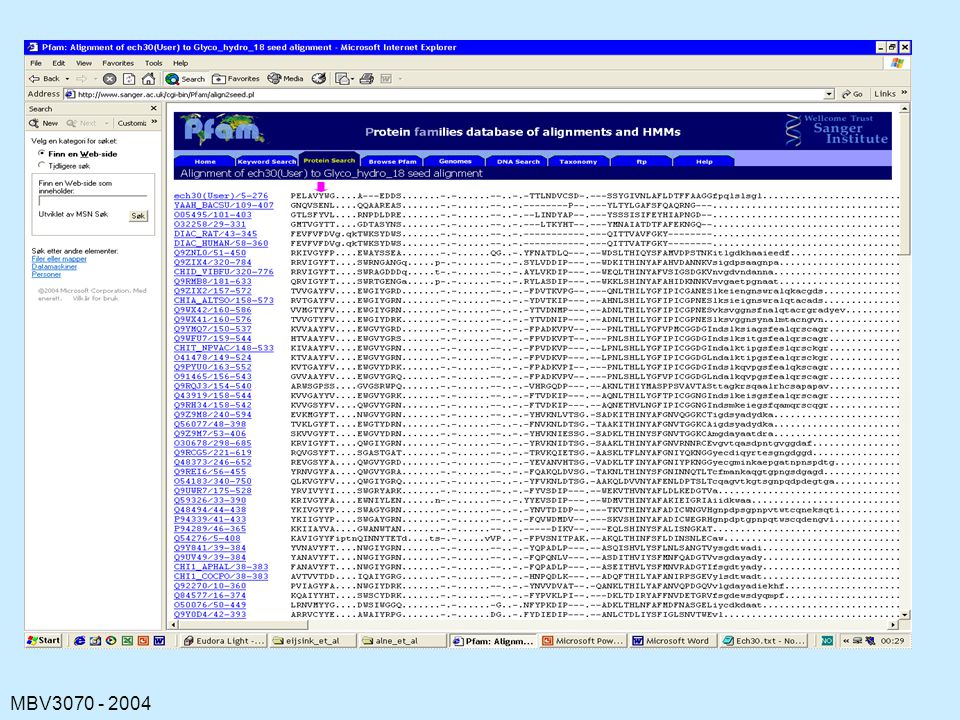
PFAM

127
PFAM

129
Aktiv sete signatur

131
Det finnes en del ”patterns with a high probability of occurrence” (som kan utelukkes i søket)
")
132
LDGFDLDnE

133
INTERPRO: Søk i mange databaser samtidig

134
Strukturelementer som er lett å predikere
Leucine zipper: sekvens hvor hver syvende aminosyre er en leucine; slike sekvenser er lett å kjenne igjen. Dette er et eksempel på en ”coiled-coil” (Nb. a og b er andre DNA-binding motiver)
")
135
Strukturelementer som er lett å predikere
Coiled coil strukturer (f eks keratin, fibrinogen): 2 eller 3 helikser som danner en supercoil; 3,5 residuer per turn (i steden for 3,6) I sekvensen ser man repeterte ”heptads” (7 residuer), hvor det 1. og 4. aminosyre er hydrofobe Det finnes programmer for å predikere coiled coils (f eks COILS)
: 2 eller 3 helikser som danner en supercoil; 3,5 residuer per turn (i steden for 3,6) I sekvensen ser man repeterte heptads (7 residuer), hvor det 1. og 4. aminosyre er hydrofobe. Det finnes programmer for å predikere coiled coils (f eks COILS)")
136
Strukturelementer som er lett å predikere: Transmembran helikser (og andre TM-områder)
Porin: b-strands

137
Prediksjon av transmembran helikser
TM-helikser er minst 19 residuer lang og nesten alle disse residuer er Hydrofobe; slike strekninger kan synliggjøres i hydropathy plots: TGREASE: hydropathy plots Nb. Det finnes forskjellige Hydrophobicity scales

138
Transmembran områder Hydropathy plots er ikke designed for å detektere transmembran områder men gir gode pekepinn. Det finnes en rekke spesialiserte programmer, f eks TMpred og PHDtopology/PHDhtm (neural netverk basert) Disse programmene er først og fremst designed for å finne transmembran helikser; de bruker mer informasjon enn bare hydrofobisitet (f eks ”stop-transfer” signaler, interface residuer osv) I de siste fem år har antallet kjente strukturer av membranproteiner steget sterkt -> dette har konsekvenser for prediktive metoder !
 Disse programmene er først og fremst designed for å finne transmembran helikser; de bruker mer informasjon enn bare hydrofobisitet (f eks stop-transfer signaler, interface residuer osv) I de siste fem år har antallet kjente strukturer av membranproteiner steget sterkt -> dette har konsekvenser for prediktive metoder !")
139
Transmembran områder TMpred

140
Transmembran områder Det er lett å finne transmembran helikser i sekvenser Man kan ofte si noe om orientering pga at man har noe som heter ”stop-transfer” signaler og en rekke andre mer eller mindre relevante kriterier (dette gjøres i PHDtopology) PHDtopology har > 90 % accuracy; feilene oppstår ved endene. OBS! proteiner som ikke har transmembranhelikser kan være assosiert med membranen allikevel (f eks gjennom ”lipid-anchors”)
 PHDtopology har > 90 % accuracy; feilene oppstår ved endene. OBS! proteiner som ikke har transmembranhelikser. kan være assosiert med membranen allikevel (f eks. gjennom lipid-anchors )")
Liknende presentasjoner










>")








-f) s. 42>")
