Laste ned presentasjonen
Presentasjon lastes. Vennligst vent

1
Studentundervisning den 24. September 2012

2
Muskelsykdommer Prevalens: ca 2-3000 i Norge som er rammet av muskelsykdom Mange forskjellige diagnoser Myopatier: patologisk process lokalisert i muskulaturen

3
Myopatier Generelle trekk: - Bilaterale symptomer - rent motoriske utfall - slappe pareser - atrofi av affisert muskulatur - svekkede (eller normale) reflekser
 reflekser")
4
Symptomene muskelsvakhet (ekstremiteter, ansikt, øye, svelg, rygg/nakke) myalgi, smerter kan forekomme ingen fascikulasjoner myotoni=forsinket relaksasjon etter kontraksjon pustevansker (svakhet i diafragma / interkostalmuskler) hjertesvikt (kardiomyopati), kardiale arytmier episodisk mørk urin (myoglobinuri)
 myalgi, smerter kan forekomme ingen fascikulasjoner myotoni=forsinket relaksasjon etter kontraksjon pustevansker (svakhet i diafragma / interkostalmuskler) hjertesvikt (kardiomyopati), kardiale arytmier episodisk mørk urin (myoglobinuri)")
5
Anamnese (1/6 spm) Arvelige muskelsykdommer i familien? Autosomal dominant Dystrophia myotonica I kr13 CTG repetisjoner, Facioscapulohumeral kr4q35, myotonia congenita, paramyotonia congenita, Autosomal recessive limb girdle muskeldystrofi, metabolske myopatier (div enzymdefekter) X-bundet (bare gutter er syke) Duchenne, Becker Andre sykdomsopphopninger (f. eks. katarakt i familien?)
 X-bundet (bare gutter er syke) Duchenne, Becker Andre sykdomsopphopninger (f. eks. katarakt i familien ).")
6
Anamnese (2/6 spm) Hvordan er forløpet? Medfødt: «floppy baby», forsinket motorisk utvikling Debutalder: Duchenne MD <5år, Myosittene i middel alder, dystrofiene i ung voksen, mitokondriepatier når som helst Akutt/subakutt: polymyositt, dermatomyositt Episodisk: metabolsk, mitokondrie, kanalopatier Monofasisk, langsom progredierende: en del av dystrofiene

7
Anamnese (3/6 spm) Har pasienten positive / negative symptomer? – Positive: smerter/myalgi (ved fysisk anstrengelse: metabolsk, mitokondrie myopati, iatrogen, inflammatoriske, thyroid), kramper (medikamentindusert, metabolsk, mitokondrie eller hypothyreoidisme), kontrakturer, stivhet (myotoni): myotonia congenita (ved aktivitet bedres), paramyotonia congenita (forverres ved aktivitet og kulde), dystrophia myotonica Hypertrofi: myotonia congenita, neuromyotonia (repetitive aktivitet), pseudohypertrofi: Duchenne, Becker – Negative: Svakhet: trappegang, reise seg fra huk, droppfot, plystring, dysartri, sover med åpne øyne fatigue atrofi
, kramper (medikamentindusert, metabolsk, mitokondrie eller hypothyreoidisme), kontrakturer, stivhet (myotoni): myotonia congenita (ved aktivitet bedres), paramyotonia congenita (forverres ved aktivitet og kulde), dystrophia myotonica Hypertrofi: myotonia congenita, neuromyotonia (repetitive aktivitet), pseudohypertrofi: Duchenne, Becker – Negative: Svakhet: trappegang, reise seg fra huk, droppfot, plystring, dysartri, sover med åpne øyne fatigue atrofi.")
8
Anamnese (4/6 spm) Triggere Anstrengelsesutløst Etter karbohydratholdig kost Bedring av hvile Temperatur Ny medikasjon
 Triggere Anstrengelsesutløst Etter karbohydratholdig kost Bedring av hvile Temperatur Ny medikasjon")
9
Anamnese (5/6 spm) Assosierte tilstander: Myoglobinuri: cola farget urin? Metabolske myopatier, fosrtyrrelse i fettstoffskifte Respiratoriske symptomer? -ortopnoe, morgenhodepine? Kardiale symptomer? – arytmia, hjertesvikt? Katarakt, mental retardasjon, skjelettdeformiteter, nevropati?

10
Anamnese / us (6/6 spm) Hvordan er paresene utbredt? Affeksjon av ansiktsmuskulatur – Dystrofia myotonica, facioscapulohumeral, oculopharyngeal dystrofier, mitokondrial Oftalmoplegi evt ptose – Mitokondrial, oculopharyngeal dystrofi. Utelukk MG! Affeksjon av proksimale muskler – Poly- dermatomyositt, limb girdle muskeldystrofi, metabolske myopatier, Hånd, finger muskler og m. quadriceps femoris – Inklusjonslegeme myositt Bare distale pareser – Dystrofia myotonica, Welander og myofibrillar myopati. Utelukk nevropati! Dysfagi, dysartri – Oculopharyngeal dystrofi, dystrofia myotonica, myositt, mitokondrial, thyroid myopati

11
Klinisk undersøkelse Kranial nerver, se etter : glatt panne, ptose, horisontalt smil, øyemuskelpareser, diplopi, dysartri, svelgproblemer, tunge (tempo, atrofi) Inspeksjon: Atrofi, hypertrofi, pseudohypertrofi,, myotonia, myokymia, Tonus (slapp/normal) Muskelkraft (prox/distale pareser, myotoni?) Refleksene Andre organ us
 Inspeksjon: Atrofi, hypertrofi, pseudohypertrofi,, myotonia, myokymia, Tonus (slapp/normal) Muskelkraft (prox/distale pareser, myotoni ) Refleksene Andre organ us")
12
Inndeling av muskelsykdommer HEREDITÆR Muskeldystrofier (defekt i strukturelle muskelproteiner) Duchenne/Becker, Facioscapulohumeral, Limb-girdle, tibial muskeldystrofi, Dystrofia myotonica, Myotonia congenita Metabolske muskelsykdommer (defekt i enzymer, stoffomsetning) mitokondrie myopati, glykogen storage disorders: Pompe, Mc'Ardles, fettstoffskifte forstyrr: carnitin mangel, Muskulære kanalopatier (dysfunksjon i ionekanaler) Hypokalemisk periodisk paralyse, hyperkalemisk periodisk paralyse, paramyotonia kongenita, myotonia kongenita
 Duchenne/Becker, Facioscapulohumeral, Limb-girdle, tibial muskeldystrofi, Dystrofia myotonica, Myotonia congenita Metabolske muskelsykdommer (defekt i enzymer, stoffomsetning) mitokondrie myopati, glykogen storage disorders: Pompe, Mc Ardles, fettstoffskifte forstyrr: carnitin mangel, Muskulære kanalopatier (dysfunksjon i ionekanaler) Hypokalemisk periodisk paralyse, hyperkalemisk periodisk paralyse, paramyotonia kongenita, myotonia kongenita")
13
Inndeling av muskelsykdommer ERVERVEDE Idiopatisk inflammatorisk muskesykdommer, Myositt (oftest autoimmun) Polymyositt, dermatomyositt, inklusjonslegeme myositt Medikament-, toksininduserte Alkohol, statiner, cicklosporin, diuretika, steroider i høye doser Endokrine Hypo- og hyperthyreose, Cushing
 Polymyositt, dermatomyositt, inklusjonslegeme myositt Medikament-, toksininduserte Alkohol, statiner, cicklosporin, diuretika, steroider i høye doser Endokrine Hypo- og hyperthyreose, Cushing")
14
Muskeldystrofier (Duchenne/Becker) X-bundet form Bare gutter affisert, ca 8-10/år i Norge Jentene er bærere (ofte lett forhøyet CK) Proximale muskel kraftsvikt Duchenne (dårlig prognose) 4/5 Debut 2-4 års alder, langsom progredierende Tidlig tenårene rullestolbrukere, ofte kardiomyopati og mental retardasjon Tidlig død ved 20 års alder Becker (bedre prognose) 1/5 Debuterer senere 7års alder-ungvoksen alder
 X-bundet form Bare gutter affisert, ca 8-10/år i Norge Jentene er bærere (ofte lett forhøyet CK) Proximale muskel kraftsvikt Duchenne (dårlig prognose) 4/5 Debut 2-4 års alder, langsom progredierende Tidlig tenårene rullestolbrukere, ofte kardiomyopati og mental retardasjon Tidlig død ved 20 års alder Becker (bedre prognose) 1/5 Debuterer senere 7års alder-ungvoksen alder")
15
Facioscapulohumoral muskeldystrofi Debut i 10-20 års alder Rammer muskulatur i ansikt, rundt skulderblad, overarm Kan være asymmetrisk og gir ofte smerter Etter hvert kan komme svakhet i ukestr og hørselstap og retinale teleangiektasier

16
Limb-girdle muskeldystrofi Symmetrsik proximal muskelaffeksjon i skulder og bekken Vanligvis autosomal recessiv men forekommer dominant form En rekke gen og proteindefekter LGMD1A (kr 5q), LGMD1B (kr 1q) LGMD2A (kr 15q – calpain-3 gen-vanligste form) LGMD2C (kr 13Q)- Ksarcoglycan gen
, LGMD1B (kr 1q) LGMD2A (kr 15q – calpain-3 gen-vanligste form) LGMD2C (kr 13Q)- Ksarcoglycan gen")
17
Dystrofia myotonica Den vanligste arvelige muskelsykdommen blant voksne En multiorgan sykdom Undertyper: DM1 – vanligst DM2 – proksimal myotonisk myopati (PROMM) DM3 – kun kasuistiske rapporter Myotoni: myotoni ved isoton kontraksjon av fingre eller perkusjon av thenar eller underarm
 DM3 – kun kasuistiske rapporter Myotoni: myotoni ved isoton kontraksjon av fingre eller perkusjon av thenar eller underarm")
18
Dystrofia myotonica Prevalens: ca 50 pr 10 6 innbyggere Autosomal dominant arvegang kromosom 19q13, (CTG)n, n>50 tripletter i DM protein kinase genet. Anticipation: alvorlighetsgraden av sykdommen øker med generasjon pga økende antall repetisjoner ved neste generasjon 50-150 mild 100-1000 klassisk DM >1000 alvorlig medfødt sykdom (kongenital) Prenatal diagnostikk er mulig
 Prenatal diagnostikk er mulig.")
19
Dystrofia myotonica Mest typiske tegn og symptomer: Symptomdebut: Ung voksen alder atrofi og pareser distalt i ekstremitetene ptose, flatt smil, halvåpen munn, skallethet, facies myotonica “misery expression” Cataract mental ret, personlighetsendringer Arytmier nedsatt kraft I resp musk, apnoe hyperinsulinemi, hypothyreoidisme

20
Merrison et al, 2009

21
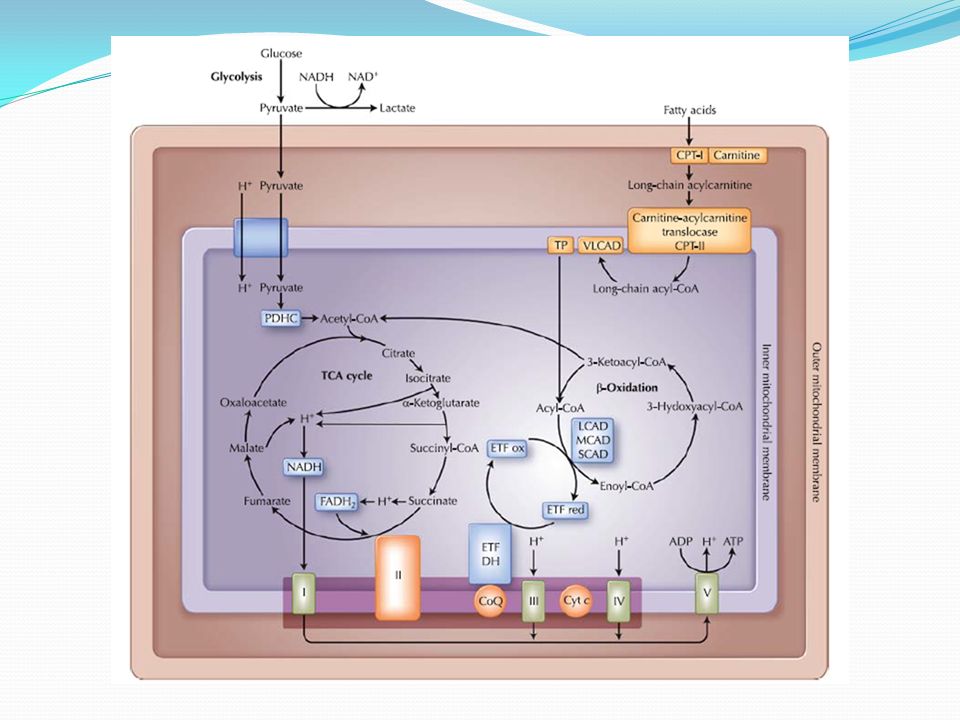
Metabolske myopatier Skyldes genetiske defekter i enzymer invovert i omsetning av karbohydrater (glykogen-glukose) eller fett og produksjon av ATP i mitokondriene De fleste er autosomalt recessive Om muskelmetabolisme: I hvile forbrenner muskler frie fettsyrer. Ved lav intensitetsbruk forbrennes både blodglukose og frie fettsyrer, ved langvarig lavintensitetsbruk nesten bare fettoksydering. Ved økende intensitet øker forbrenning av glykogen. fysisk aktivitet kan gi smerter, hevelse og ømhet i muskulaturen, ofte med utskillelse av myoglobin i urin

23
Inndeling av metabolske myopatier Myopatier som skyldes defekt i karbohydratstoffskiftet (glykogenosene) Acid maltase mangel (Pompe) Glucose-6-phosphatase mangel Debrancher enzyme mangel (Cori-Forbes disease) Brancher enzyme mnagel (Andersen disease) muskel phosphorylase mangel (McArdle disease) Phosphofructokinase mangel (Tarui disease) Myopatier som skyldes defekt fettstoffskiftet Karnitin Palmitoyl Transferase mangel, Karnitin mangel Mitokondrie myopatier isolert myopati eller ledd i mer omfattende syndrom (MERFF, MELAS etc)
 Acid maltase mangel (Pompe) Glucose-6-phosphatase mangel Debrancher enzyme mangel (Cori-Forbes disease) Brancher enzyme mnagel (Andersen disease) muskel phosphorylase mangel (McArdle disease) Phosphofructokinase mangel (Tarui disease) Myopatier som skyldes defekt fettstoffskiftet Karnitin Palmitoyl Transferase mangel, Karnitin mangel Mitokondrie myopatier isolert myopati eller ledd i mer omfattende syndrom (MERFF, MELAS etc)")
24
Acid maltase mangel (Pompe sykdom) Autosomal recessive glykogenlagringssykdom Defekt i enzymet alfa glycosidase (GAA) Infantil form: de fleste dør før 2-års alder Adult form: kan presentere seg når som helst etter 20 års alder langsomt progredierende proximale pareser og atrofi Respirasjonssvikt er vanlig og en del presenterer seg med det. Økt forekomst av basilarisaneurismer. Muskel biopsi kan vise glykogen vakuoler Enzym måling (alfa glycosidase) i muskel, lymfocytter, eller hudfibroblaster
 i muskel, lymfocytter, eller hudfibroblaster.")
25
Myofosforylase mangel (mc Ardle sykdom) Autosomal recessiv, debuterer ofte før 20 års alder Muskelbruk utløser smertefull hevelse, tretthet, eller svakhet, varer noen timer og lokaliseres til brukte muskler Bedrer seg igjen etter en tids bruk ("Second wind") når fettsyre-forbrenning dominerer Klinisk us er som oftest normal, men noen får muskelhypertrofi (24%) og noen får permanente proksimale pareser og mild atrofi 50% har myoglobinuri, alle har høy CK Diagnostikk: klinikk og enzymmåling (myophosphorylase) i muskel
 Autosomal recessiv, debuterer ofte før 20 års alder Muskelbruk utløser smertefull hevelse, tretthet, eller svakhet, varer noen timer og lokaliseres til brukte muskler Bedrer seg igjen etter en tids bruk ( Second wind ) når fettsyre-forbrenning dominerer Klinisk us er som oftest normal, men noen får muskelhypertrofi (24%) og noen får permanente proksimale pareser og mild atrofi 50% har myoglobinuri, alle har høy CK Diagnostikk: klinikk og enzymmåling (myophosphorylase) i muskel")
26
Carnitin palmitoyltransferase mangel (fettsyrestoffskifte) Kan presentere seg hos voksne (helst menn). Debuterer ofte med rhabdomyolyse og myoglobinuri - kan gi nyresvikt (mangler hos 20%) Episodiske anstrengelsesutløste symptomer; muskelsmerter, stivhet og ømhet men sjelden kramper. Smertene kommer ofte etter aktivitet. Anfallene trigges av lang aktivitet særlig ved faste eller kaldt vær, søvnmangel, febersykdom, emosjonelt stress. Normal CK mellom anfall. Normal muskelbruktest, Normal EMG, Muskelbiopsi kan være normal, evt lipiddråper subsarkolemmalt, dg: blodprøve til acylcarnitin us
 Episodiske anstrengelsesutløste symptomer; muskelsmerter, stivhet og ømhet men sjelden kramper. Smertene kommer ofte etter aktivitet. Anfallene trigges av lang aktivitet særlig ved faste eller kaldt vær, søvnmangel, febersykdom, emosjonelt stress. Normal CK mellom anfall. Normal muskelbruktest, Normal EMG, Muskelbiopsi kan være normal, evt lipiddråper subsarkolemmalt, dg: blodprøve til acylcarnitin us.")
27
Mitokondriepatier knyttet til organismens energiomsetning. Alle kroppens organer og funksjoner er avhengige av energi i vekslende grad og mitokondriesykdommer er karakterisert ved sammensatte sykdomsbilder fra flere organsystemer Mitokondriesykdommer kan: affisere alle vev presenteres i alle aldre ha alle slags arvemønstre

28
Mitokondriepatier Mitokondrie-encefalopati, laktacidose & "strokelike" episodes (MELAS): Gir hodepine som kan ligne migrene med kvalme og ubehag, illebefinnende med nevrologiske tegn som skyldes forbigående eller mer varige sirkulasjonsforstyrrelser i hjernen (slag-lignende). Epilepsi som kan være vanskelig å kontrollere. Myopati, encefalopati, "ragged red fibers" (MERRF): Gir muskelsvakhet, myoklonus, epilepsi, syns- og hørselshemning, motoriske vansker og lærevansker.
: Gir muskelsvakhet, myoklonus, epilepsi, syns- og hørselshemning, motoriske vansker og lærevansker..")
29
Kanalopatier/periodiske paralyser De best kjente og hyppigste blant periodiske pareser er hypokalemisk / hyperkalemisk periodisk parese Autosomal dominant arvegang med lav penetrans som fører til defekt av ionekanaler Klinisk bilde: Periodisk paralyse Myotoni (EMG) Malign hypertermi (ved ulike anestesimidler) Anfallsvise pareser med unormal kaliumnivå under anfall, karbohydratrik mat kan utløse
 Malign hypertermi (ved ulike anestesimidler) Anfallsvise pareser med unormal kaliumnivå under anfall, karbohydratrik mat kan utløse")
30
Inflammatoriske myopatier Subtyper: Polymyositt Dermatomyositt Inklusjonslegeme myositt Relativt akutt/subakutt start Angivelig autoimmun etiologi, komplikasjon til RA, SLE Malignitetsutredning viktig!! (ovarie, GI, mamma, lunge lever) Proksimale muskler involvert
 Proksimale muskler involvert.")
31
IBM mønster

32
Drug induced muscle problems include Asymptomatic raised plasma CK (statins) Myalgia and cramps (statins, diuretics) Myotonia (chloroquine, beta 2 adrenergic blockers) Necrotising myopathy/rhabdomyolysis (statins, cocaine) Chronic progressive myopathy (corticosteroids) Rarely mitochondrial myopathy (AZT treatment in HIV) or inflammatory myopathy (D-penicillamine) may be drug- induced.
 Myalgia and cramps (statins, diuretics) Myotonia (chloroquine, beta 2 adrenergic blockers) Necrotising myopathy/rhabdomyolysis (statins, cocaine) Chronic progressive myopathy (corticosteroids) Rarely mitochondrial myopathy (AZT treatment in HIV) or inflammatory myopathy (D-penicillamine) may be drug- induced.")
33
Merrison et al, 2009.

34
Diagnostikk 2, Lab analyser serum CK (s-creatin kinase): Oftest forhøyet men kan være normal: ved enkelte metabolske myopatier, kongenitale myopatier, utbrendte myositter Kan være forhøyet ved enkelte andre tilstander: Etter fysisk anstrengelse, traume, sepsis, hypotermi, hjertesykdom Sykdommer med denervering men som regel <1000IU/l Jo høyere CK jo mer aktiv muskelnekrose
: Oftest forhøyet men kan være normal: ved enkelte metabolske myopatier, kongenitale myopatier, utbrendte myositter Kan være forhøyet ved enkelte andre tilstander: Etter fysisk anstrengelse, traume, sepsis, hypotermi, hjertesykdom Sykdommer med denervering men som regel <1000IU/l Jo høyere CK jo mer aktiv muskelnekrose")
35
Diagnostikk Nevrofysiologi (EMG og nevrografi) Normal nevrografi EMG: ofte typiske forandringer (små, korte, polyfasiske motor unit potensialer og tidlig tett interferens (recruitment) Myotone utladninger finnes ved dystrofia myotonika og andre myotonier, Pompe’s sykdom, og hypothyroidisme kan være normal ved metabolske myopatier
 Normal nevrografi EMG: ofte typiske forandringer (små, korte, polyfasiske motor unit potensialer og tidlig tett interferens (recruitment) Myotone utladninger finnes ved dystrofia myotonika og andre myotonier, Pompe’s sykdom, og hypothyroidisme kan være normal ved metabolske myopatier")
37
Diagnostikk Muskelbiopsi: Valg av rett muskel er viktig. Ved langvarige symptomer bør en velge moderat svak muskel. Ved kort varighet bør en velge svakeste muskel. Patologien kan være fokal/flekkvis og fanges ikke alltid opp ved en enkelt biopsi.

38
Sendes til patologen direkte uten tilsatt av noe

39
Dystrofinopati - immunhistokjemi NormalDuchenne

40
Red ragged fibers – Mitokondriepatier MERRF

41
Muskelbiopsi ved myosittene Typiske inflammatoriske forandringer med t- lymfocytter og makrofager Ved dermatomyositt: perifascikulær atrofi Ved IBM: vacuoler og usp inflammasjon

42
Diagnostikk MR av muskler kan detektere subkliniske forandringer (særlig dype muskler) og mønstre som kan være diagnostisk nyttig Kan være nyttig for å finne egnet muskel for biopsi hvis den første var negativ
 og mønstre som kan være diagnostisk nyttig Kan være nyttig for å finne egnet muskel for biopsi hvis den første var negativ")
43
Diverse blodprøver Ca 60% av myositt pasientene har en myositt spesifikt antistoff: Anti-Jo -1: deramtomyositt Anti-signal recognition particle (SRP): dårlig respons på beh. Anti-Mi2: klassisk dermatomyositt Antisyntetase antistoffer: anti-KS, OJ, EJ, PL-7, PL-12, U1-RNP Sendes til RH De andre til Lund

44
Andre prøver – antistoffer i serum ved myositter. Link til rekvisisjonmyositterLink til rekvisisjon – acylcarnitin i serum og plasma ved mistanke om carnitin palmitoyl transferase mangel. Link til rekvisisjon carnitin palmitoyl transferase mangel Link til rekvisisjon – laktat og amoniakk i plasma/serum, evt etter muskelbruk (se muskelbruktest) ved mistanke om enkelte metabolske muskelsykdommer og mitokondriesykdommermuskelbruktestmetabolske muskelsykdommer mitokondriesykdommer – organiske syrer i serum og urin, og spesifikke enzymer i muskel, blod, urin på klinisk mistanke.
 ved mistanke om enkelte metabolske muskelsykdommer og mitokondriesykdommermuskelbruktestmetabolske muskelsykdommer mitokondriesykdommer – organiske syrer i serum og urin, og spesifikke enzymer i muskel, blod, urin på klinisk mistanke..")
45
Andre tester Ved myosittene er det viktig med malignitetsutredning (Dermatomyositt utvikler ca de neste 2-3 årene: ovarie, GI, mamma, lunge lever) Generelt MG med tensilontest
 Generelt MG med tensilontest")
46
Genetikk www.genetikkportalen.no

Liknende presentasjoner








>")









 Nevrofibromatose type 2 (NF2)>")
