Laste ned presentasjonen
Presentasjon lastes. Vennligst vent

1
HLH Hemofagocytisk lymfohistiocytose Magnhild Eide Macpherson
LIS, Rikshospitalet

2
Hva er HLH? Et syndrom med patologisk immun aktivering
Familiær eller sporadisk Mange mulige triggere Cytopenier Ekstrem inflammasjon Vanskelig å diagnostisere (tidlig nok) Rask oppstart av immunkjemoterapi essensielt for overlevelse
 Rask oppstart av immunkjemoterapi essensielt for overlevelse.")
3
Bakgrunn 1952: familiær hemofagocytisk reticulose
Familiær immundysregulatorisk sykdom hos barn Senere: både familiær sykdom og sporadiske tilfeller Assosiert med infeksjoner malignitet reumatologisk sykdom (MAS) Cytotoxiske mangler og øvrig affeksjon av immunsystemet 1999: Perforin mutasjoner hos enkelte HLH pasienter
 Cytotoxiske mangler og øvrig affeksjon av immunsystemet. 1999: Perforin mutasjoner hos enkelte HLH pasienter.")
4
Dyrestudier: cytotoxiske mangler fører til
unormal T-celle aktivering og inflammatorisk cytokin produksjon : driver sykdoms progresjon

5
Helgenom ekspresjon analyser:
paradoksal nedregulering av immunresponsen inkl. B-celle utvikling og funksjon TLR ekspresjon og signalering Apoptose induksjon

6
Diagnose Gjenkjenne en unik sammensetning av kliniske funn
Undersøke sykdoms assosierte genetiske defekter Bekrefte klinisk diagnose Risiko for fremtidig tilbakefall av sykdommen Definerer HLH predisposisjon hos asymptomatiske familiemedlemmer Diagnostiske kriterier basert på retrospektive analyser av pasienter kategorisert med og behandlet for HLH over flere tiår HLH-94 kriteriene; som ledd i en klinisk studie formulerte The Histiocyte Society (USA) i 1994 en standard definisjon for HLH, revidert i 2004
 i 1994 en standard definisjon for HLH, revidert i")
7
Diagnosekriterier HLH-04 studien:
A: Molekylær diagnose forenlig med HLH Patologisk mutasjon av PRF1, UNC13D, Munc18-2, Rab27a, STX11, SH2D1A eller BIRC4 B: Fem av åtte kriterier som listet under: 1 Feber ≥38,5 C 2 Splenomegali 3 Cytopenier (affiserer minst 2 av 3 cellelinjer i perifert blod) Hb < 9 g/dl Trbc < 100 x 103/ml Nøytrofile < 1 x 103/ml 4 Hypertriglyseridemi og/eller lavt fibrinogen 5 Hemofagocytose i benmarg, milt, lymfeknuter eller lever 6 Lav eller manglende NK-celle aktivitet 7 Ferritin > 500 ng/ml 8 Forhøyet sCD25 (alpha-kjede av løselig IL-2 reseptor)
 Hb < 9 g/dl. Trbc < 100 x 103/ml. Nøytrofile < 1 x 103/ml. 4 Hypertriglyseridemi og/eller lavt fibrinogen. 5 Hemofagocytose i benmarg, milt, lymfeknuter eller lever. 6 Lav eller manglende NK-celle aktivitet. 7 Ferritin > 500 ng/ml. 8 Forhøyet sCD25 (alpha-kjede av løselig IL-2 reseptor)")
8
Diagnose Ved familiær HLH bør det i tillegg ikke foreligge tegn til malignitet Selv om HLH-04 protokollen bruker ferritin > 500, anses ferritin > 3000 som bekymringsverdig mtp HLH og verdier > svært sensitivt og spesifikt for HLH NB: ikke alle har hemofagocytose ved diagnosetidspunktet, derfor er diagnosen ikke avhengig av dette funnet alene sCD25 (løselig IL-2 reseptor) en viktig inflammasjons markør; korrelerer mer konsistent med sykdomsaktivitet enn ferritin
 en viktig inflammasjons markør; korrelerer mer konsistent med sykdomsaktivitet enn ferritin.")
9
Diagnose Kriteriene reflekterer ikke alle typiske kliniske funn eller laboratoriefunn ved HLH Mange har lever betennelse (mild grad til leversvikt) Uforklart leversvikt, cytopenier og høye inflammasjonsparametre bør trigge at man tenker på HLH HLH diagnose med normale leverprøver vil være uvanlig

10
Diagnose Nevrologiske symptom er relativt vanlige
Distinkt klinisk trekk hos mange med familiær HLH patogenese En rekke HLH assosierte molekylære markører er ikke med i kriteriene, men kan nå analyseres Proteiner (perforin, SLAM-associated protein, X-linked inhibitor of apoptosis protein) Overflate CD107a (indikerer genetisk avvik som affiserer degranulering)
 Overflate CD107a (indikerer genetisk avvik som affiserer degranulering)")
11
Patofysiologi I PREDISPONERENDE IMMUNSVIKT:
Lav eller manglende NK-celle funksjon Genetisk defekt som affiserer cytotoxisitet Familieanamnese på HLH Tidligere episode(r) med HLH eller uforklart cytopeni Markører på manglende cytotoxisitet Redusert ekspresjon av perforin, SAP, XIAP Mobilisering av CD107a
 med HLH eller uforklart cytopeni. Markører på manglende cytotoxisitet. Redusert ekspresjon av perforin, SAP, XIAP. Mobilisering av CD107a.")
12
Patofysiologi II SIGNIFIKANT IMMUNAKTIVERING: Feber
Splenomegali / hepatomegali Forhøyet ferritin (> 3000 ng/ml) Forhøyet sCD25 Forhøyet sCD16393 Ved behandling av HLH ser man at ferritin og sCD25 ofte stiger før man ser klinisk forverring av HLH sCD163 økt ved inflammasjon og makrofag aktivering
 Forhøyet sCD25. Forhøyet sCD Ved behandling av HLH ser man at ferritin og sCD25 ofte stiger før man ser klinisk forverring av HLH. sCD163 økt ved inflammasjon og makrofag aktivering.")
13
Patofysiologi III UNORMAL IMMUNPATOLOGI: Cytopenier
Lav fibrinogen eller høye triglyserider Hemofagocytose Hepatitt CNS involvering

14
Patofysiologi Eksperimentelle studier: normal cytotoxisk funksjon begrenser immunaktivering og reduserer slik utvikling av alvorlig immunpatologi Underliggende immunaktivering ved HLH utvikling Ses ofte før hemofagocytose evt utvikles Kombinasjonen av akutt systemisk immunaktivering og unormal immunpatologi er det som skiller HLH fra andre inflammatoriske sykdommmer

15
Organspesifikke infeksjoner eller autoimmune prosesser (f
Organspesifikke infeksjoner eller autoimmune prosesser (f.eks hepatitt, meningitt, aplastisk anemi) gir som regel ikke samme systemiske inflammasjon / multiorgansvikt Systemiske inflammatoriske prosesser (f.eks sepsis) gir som regel ikke den samme spesifikke konstellasjonen av immunpatologi og T-celle aktivering Ved diagnose: bør finne kriterier både på immunaktivering og unormal immunpatologi
 gir som regel ikke samme systemiske inflammasjon / multiorgansvikt. Systemiske inflammatoriske prosesser (f.eks sepsis) gir som regel ikke den samme spesifikke konstellasjonen av immunpatologi og T-celle aktivering. Ved diagnose: bør finne kriterier både på immunaktivering og unormal immunpatologi.")
16
Primær vs sekundær HLH Primær Familieanamnese, genetiske årsaker
Vanligvis spedbarn / barn Defekt cytotoxisk funksjon (ikke alltid) Risiko for tilbakefall, trenger som regel HCT Immunologisk trigger ofte ikke opplagt Sekundær Trigger: infeksjon (EBV), malignitet, revmatol. sykdom Mindre sjanse for tilbakefall, eldre barn og voksne Begrenset verdi av å dele inn i primær / sekundær
 Risiko for tilbakefall, trenger som regel HCT. Immunologisk trigger ofte ikke opplagt. Sekundær. Trigger: infeksjon (EBV), malignitet, revmatol. sykdom. Mindre sjanse for tilbakefall, eldre barn og voksne. Begrenset verdi av å dele inn i primær / sekundær.")
17
MAS – Macrophage Activation Syndrome
Den viktigste, potensielt dødelige komplikasjonen til systemisk juvenil idiopatisk artritt Også assosiert med SLE : HLH assosiert med revmatologisk sykdom Feber, hepatosplenomegali, hepatitt, lymfadenopati, DIC Cytopenier kommer sent i forløpet (høye trbc og nøytr pga revma sykdom) Hemofagocytose (benmarg, annet vev) viktig funn
 Hemofagocytose (benmarg, annet vev) viktig funn.")
18
MAS Infeksjoner, oppbluss av systemisk juvenil idiopatisk artritt, endring av medisiner kan utløse MAS Immunologisk og genetisk nær relasjon til HLH De fleste har redusert NK-celle funksjon, redusert ekspresjon av perforin, forhøyet sCD25 og sCD163 Behandling med økt immunsuppresjon og høydose IVIG er ofte effektivt Hos noen effekt av Kineret og Roactemra (anti IL-6) Ved forverring tross steroider, cyclosporin el. øvrig sykdoms-spesifikk behandling: aktuelt med etoposide, annen HLH salvage terapi, HCT
 Ved forverring tross steroider, cyclosporin el. øvrig sykdoms-spesifikk behandling: aktuelt med etoposide, annen HLH salvage terapi, HCT.")
19
HLH trigger: malignitet
Primært ved lymfom eller NK-/T-celle leukemi Også ved andre leukemier og solide tumores Mange har simultant en infeksjon (bakt./viral/sopp) Ved diagnosetidspunkt: CT thorax / abdomen, benmargsaspirat og –biopsi mtp identifisere evt underliggende malignitet Ved malignitet: anbefales primært å starte immunkjemoterapi for å kontrollere inflammasjon, deretter sykdoms-spesifikk behandling når inflammasjons markørene har normalisert seg
 Ved diagnosetidspunkt: CT thorax / abdomen, benmargsaspirat og –biopsi mtp identifisere evt underliggende malignitet. Ved malignitet: anbefales primært å starte immunkjemoterapi for å kontrollere inflammasjon, deretter sykdoms-spesifikk behandling når inflammasjons markørene har normalisert seg.")
20
HLH trigger: EBV EBV er den hyppigste infeksjonen assosiert med HLH
EBV-HLH assosiert med akutte infeksjoner i B-celler, men også i T- og NK-celler Bedre overlevelse hvis etoposide er del av behandlingen, og rask oppstart etter diagnose Rituximab kan være nyttig fordi den kan eliminere EBV-infiserte B-celler hos de med progressiv EBV-HLH Noen pasienter som har tilsynelatende selvlimiterende HLH etter primær EBV infeksjon vil senere utvikle aggressiv residiverende HLH som de trenger immun-kjemoterapi og HCT for

21
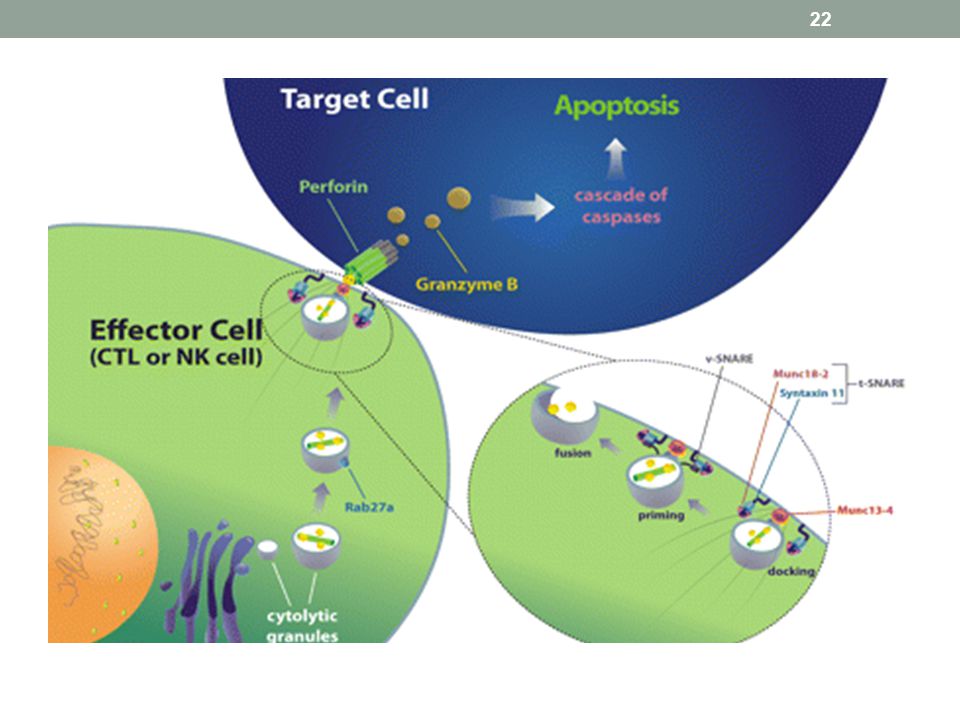
Genotype og fenotype Flere kjente genetiske syndrom assosiert med HLH
Alle fører til den samme fenotypen med svekket cytotoxisk funksjon hos T- og NK-celler og predisposisjon for å utvikle HLH HLH assosierte genetiske variasjoner kan affisere granula-avhengig lymfocytt cytotoxisitet ved å svekke trafficking, docking, priming for exocytose eller membran fusjon av cytotoxiske granulae. Tap av funksjonelt perforin

23
Kliniske aspekt ved HLH
Langvarig feber Hos pasienter med FUO, cytopenier, ferritin > 3000 eller signifikant forhøyet sCD25: gjør HLH diagnostikk Leversykdom og koagulopati HLH bør være diff.diagnose ved akutt leversvikt, særlig ved lymfocytiske infiltrat på leverbiopsi Nærmere 95% av pasientene har trekk av DIC og er høyrisiko for akutte blødninger Noen har blodplate dysfunksjon

24
Kliniske aspekt ved HLH
Benmargssvikt: Anemi og trombocytopeni hos >80% ved presentasjon Hemofagocytose hos % ved diagnose NB: Hemofagocytose i benmarg kan ses uten at HLH foreligger Blodtransfusjoner, infeksjoner, autoimmun sykdom, RBC destruksjon Infiltrasjon i benmarg av aktiverte makrofager, sammen med klinisk vurdering, kan skille HLH fra andre årsaker til hemofagocytose

25
Kliniske aspekt ved HLH
Hudmanifestasjoner: Hos 6-65%, svært varierende hudutslett Utslett kan korrelere med lymfocytt infiltrasjon på biopsi, og hemofagocytose kan også ses. Ta hudbiopsi ved utredning! Lungesykdom Forverring respiratorisk er et svært dårlig tegn, indikerer dårlig HLH kontroll og/eller infeksjon CNS, oftalmologisk og nevromuskulær affeksjon Over 1/3 av pasientene har nevrologiske symptom i starten CSF er unormal hos >50% av HLH pasienter (pleocytose, høyt protein, og/eller hemofagocytose)
")
26
Behandling av HLH Uten behandling: overlevelse ved aktiv F-HLH ca 2 mnd HLH-94 protokollen: -8 uker induksjonsterapi med dexamethasone, etoposide og intrathekal MTX. -Behandlingsmål: slå ned inflammatorisk prosess. -Etter 8 uker: gradvis seponering av behandling eller overgang til kontinuerings terapi (som bro til benmargstransplantasjon)
")
27
Behandling av HLH HLH-2004 studien inkluderer fortsatt pasienter: sammenlignet med tidligere behandling ble cyklosporin flyttet til begynnelsen av induksjonsbehandlingen, hydrokortison lagt til som intrathekal behandling Single-centre publikasjon basert på erfaring over 14 år: som alternativ til etoposid-holdige regimer har de gitt kortikosteroider og ATG, raskt etterfulgt av HCT (trenger videre studier)
")
28
Induksjonsterapi Start raskt, tross evt infeksjoner som ikke er ferdigbehandlet, cytopenier eller organ dysfunksjon HLA typing sendes ved start av induksjonsterapi Kan være uforutsigbart forløp; må ofte individuelt tilpasse behandlingen Dexametason dose og etoposide hyppighet av dosering må iblant økes ved reaktivering av sykdom

29
Induksjonsterapi Dersom pasienten ikke viser i noe respons på behandlingen ila 2-3 uker, bør man vurdere salvage terapi Økende feber og inflammasjonsmarkører etter behandlingsrespons: tenk på opportunistiske infeksjoner

30
Initiale vurderinger Sett diagnosen tidlig nok!
Søk etter triggere; start evt spesifikk antimikrobiell terapi Stabil pasient, ikke veldig syk: kan vurdere å behandle underliggende tilstand primært, evt samtidig med kortikosteroider, tett oppfølging Rituximab kan være nyttig ved EBV-drevet HLH IVIG ved virale infeksjoner Økende transaminaser, bilirubin, ferritin, koagulopati, s-IL2-r nivå og dårligere respiratorisk status; dårlige prognostiske tegn

31
CNS sykdom Krampeanfall, endret bevissthet, facialisparese, dysartri, dysfagi, pleocytose i CSF Spinalpunksjon og MR caput hører med, selv hos asymptomatiske Ved CNS affeksjon: ukentlig IT-MTX og hydrocortison til CSF normaliseres og symptomene klinger av Risiko for posterior reversibel encephalopati syndrom ila induksjons behandling (obs ved HTN); aggressiv BT beh. CNS affeksjon indikerer primær HLH; økt risiko for langtids morbiditet: vurder HCT
; aggressiv BT beh. CNS affeksjon indikerer primær HLH; økt risiko for langtids morbiditet: vurder HCT.")
32
Støttende behandling Infeksjonsprofylakse som hos øvrige benmargstransplanterte Høy risiko for blødning: anbefaler ikke profylaktisk heparin. Holde trbc >50. Vær obs på akutt kardial dysfunksjon, ekko cor

33
Videre behandling HCT anbefalt hos de med CNS affeksjon, refraktær eller residiverende sykdom, persisterende NK-celle dysfunksjon, familiær type Etter induksjon gis pulsbehandling med Dexametason og etoposid. Kan legge til ciklosporin Pasienter på kontinuerings behandling bør gå til HCT så raskt som mulig Forsøkt ATG og Alemtuzumab (anti-CD52) som salvage terapi
 som salvage terapi.")
34
HCT ved HLH Langtids sykdomsfri overlevelse etter HCT 50-65% før år 2000 Japansk studie: 80% langtidsoverlevelse hos 14 pasienter som ble transplantert for EBV-assosiert HLH RIC (redusert intensitet conditioning): de fleste RIC forbehandling regimer inkluderer alemtuzumab og viser bedre overlevelse tidlig etter transplantasjon NB: donorvalg. Occult predisponering for HLH dersom søsken donor er mulig Full donor kimerisme post-Tx ikke nødvendig for å undertrykke HLH sykdom hos de fleste
: de fleste RIC forbehandling regimer inkluderer alemtuzumab og viser bedre overlevelse tidlig etter transplantasjon. NB: donorvalg. Occult predisponering for HLH dersom søsken donor er mulig. Full donor kimerisme post-Tx ikke nødvendig for å undertrykke HLH sykdom hos de fleste.")
35
Oppfølging etter behandling
Mange av de som får residiv av sykdommen får det ila 1 år etter behandling Månedlige kontroller det første året Deretter årlige kontroller

Liknende presentasjoner















 Nevrofibromatose type 2 (NF2)>")




